
Abstract
Xenotransplantation of genetically engineered porcine chondrocytes may benefit many patients who suffer cartilage defects. In this work, we sought to elucidate the molecular bases of the cellular response to xenogeneic cartilage. To this end, we isolated pig costal chondrocytes (PCC) and conducted a series of functional studies. First, we determined by flow cytometry the cell surface expression of multiple immunoregulatory proteins in resting conditions or after treatment with human TNF-α, IL-1α, or IL-1β, which did not induce apoptosis. TNF-α and to a lesser extent IL-1α led to a marked upregulation of SLA I, VCAM-1, and ICAM-1 on PCC. SLA II and E-selectin remained undetectable in all the conditions assayed. Notably, CD86 was constitutively expressed at moderate levels, whereas CD80 and CD40 were barely detected. To assess their function, we next studied the interaction of PCC with human monoblastic U937 and Jurkat T cells. U937 cells adhered to resting and in a greater proportion to cytokine-stimulated PCC. Consistent with its expression pattern, pig VCAM-1 was key, mediating the increased adhesion after cytokine stimulation. We also conducted coculture experiments with U937 and PCC and measured the release of pig and human cytokines. Stimulated PCC secreted IL-6 and IL-8, whereas U937 secreted IL-8 in response to PCC. Finally, coculture of PCC with Jurkat in the presence of PHA led to a marked Jurkat activation as determined by the increase in IL-2 secretion. This process was dramatically reduced by blocking pig CD86. In summary, CD86 and VCAM-1 on pig chondrocytes may be important triggers of the xenogeneic cellular immune response. These molecules together with TNF could be considered potential targets for intervention in order to develop xenogeneic therapies for cartilage repair.
Introduction
Cartilage is an avascular tissue with little spontaneous regenerative capacity (30,37). The consequent great medical needs and the prospect of better therapies has promoted intense research in cartilage tissue engineering and transplantation. The repair of articular cartilage defects is being attained in a limited number of patients by autologous transplantation of chondrocytes (30,37). Despite this success, allotransplantation is being considered as an alternative to autologous grafting for settings that require whole menisci, large number of cells, and/or when autologous chondrocytes are difficult to obtain (24,38). Nasal, auricular, and tracheal reconstructions are other indications in need of a good source of chondrocytes/cartilage. Research in stem cell technology may provide a future solution to this problem (15,25). However, we believe xenotransplantation may be another new interesting option for cartilage repair that is worth exploring. Xenotransplantation offers an unlimited supply of cells that can be obtained under very high-quality standards (1,14). Xenogeneic chondrocytes could as well be selected based on their regenerative capacity and, most importantly, the xenogeneic donor cells and tissues can be adapted by genetic engineering to the intended clinical applications (1).
Tissues of bovine and porcine origin have being extensively used as basic raw materials in tissue engineering research (31). However, the rejection process of tissue-based xenografts needs to be further elucidated in order to develop strategies that allow engraftment in a clinical setting. Porcine cartilage transplanted into primates is rejected within weeks to months by a process of tissue rejection comprising humoral and cellular responses (33). As a first barrier, human natural antibodies bind to porcine chondrocytes by mainly recognizing the carbohydrate antigen Gal α1,3-Gal (Gal) expressed in most pig tissues (8). However, these antibodies do not cause hyperacute rejection of the avascular tissue. Transplantation of pig cartilage also induces high titers of anti-Gal and non-Gal antibodies in monkeys (32,33). The anti-Gal response plays a major role as removal of the Gal epitope by α-galactosidase treatment before implantation markedly reduced the elicited anti-Gal antibody response and the amount of cellular immune infiltrate in the xenograft (32). We further confirmed a key role of the Gal antigen in promoting rejection, as transgenic expression of H transferase (HT) in porcine cartilage markedly reduced Gal antigen expression and averted the anti-Gal antibody response induced after transplantation into α1,3-galactosyltransferase-deficient mice (Gal KO mice, which lack the Gal epitope) (8). Moreover, HT-transgenic grafts led to a milder anti-pig antibody response and showed a diminished cellular immune infiltrate (8).
The cellular immune infiltrate of pig cartilage xenografts 2 months after transplantation in cynomolgus monkeys contains 80–90% of T cells (half CD4+ and half CD8+) and 10–20% macrophages (33). In accordance with the cellular rejection process of other pig xenografts such as pancreatic islets and skin (36,41), both CD4+ and CD8+ T cells contribute to cartilage rejection. Systemic depletion of CD4+ T cells in a pig-to-mouse cartilage transplant model (in the absence of an anti-Gal response) reduced both T cells and macrophages in the xenograft (8). Although rejection was delayed, it still proceeded by a mononuclear cellular infiltrate. In general, the cellular immune response plays a critical role in rejection of cell-based xenografts. The long-term graft survival observed in preclinical primate models of pancreatic islet transplantation using intensive immunosuppression supports this view (4). However, the underlying mechanisms remain unknown, especially for rejection of pig cartilage. A better understanding of the cellular responses triggered by xenogeneic cells and identifying the key molecules involved would certainly help to develop the strategies needed for clinical success.
In this work, we sought to elucidate the molecular bases of cell-mediated rejection of xenogeneic cartilage. To this end, we isolated pig costal chondrocytes (PCC) and conducted a series of functional studies that included treatment with human inflammatory cytokines and xenogeneic cell-based assays. These experiments allowed us to identify cytokines, adhesion and costimulatory molecules that may be considered targets for intervention in cartilage xenotransplantation.
Materials and Methods
Isolation and Culture of Porcine Cells
PCC were isolated from pig costal cartilage according to previously published methods (40). Briefly, costal cartilage was dissected from the ribs after euthanasia under sterile conditions following animal procedures approved by the local ethical committee. Cartilage was cut into small pieces washed in PBS supplemented with antibiotics at 4°C and subsequently treated with 0.8% w/v pronase (Sigma, St. Louis, MO) for 1 h at 37°C with stirring. The enzymatic digestion was continued for 8 h more by addition of 0.4% type II collagenase (Invitrogen, Carlsbad, CA). Tissue was collected by centrifugation, sieved with a 100-μm nylon mesh (BD Biosciences, San Jose, CA), washed, and resuspended in Ham's F12 (Invitrogen)/20% FBS (Biological Industries, Kibbutz Beit Haemek, Israel) supplemented with penicillin, streptomycin, and gentamicin (200 U/ml, 200 and 50 μg/ml, respectively, Invitrogen). Cells were seeded into tissue culture flasks and maintained as monolayer in Ham's F12/DMEM (1:1) or DMEM (Invitrogen) supplemented with 10% FBS and 25 mg/L endothelial mitogen (EM, Biomedical Technologies, Stoughton, MA). EM was withdrawn 24 h before carrying out the functional assays to avoid interference. For cytokine treatments, PCC were incubated for 24 h with either human tumor necrosis factor-α (hTNF-α), human interleukin-1α (hIL-1α), or hIL-1β (Peprotech EC, London, UK) at 10 ng/ml in culture medium without EM. Porcine aortic endothelial cells (PAEC) were obtained from European Collection of Cell Cultures (Porton Down, UK) and cultured in DMEM/10% FCS. The culture medium was supplemented with EM (50 mg/L) for cell expansion, but withdrawn 24 h prior to assays.
Characterization of PCC by RT-PCR
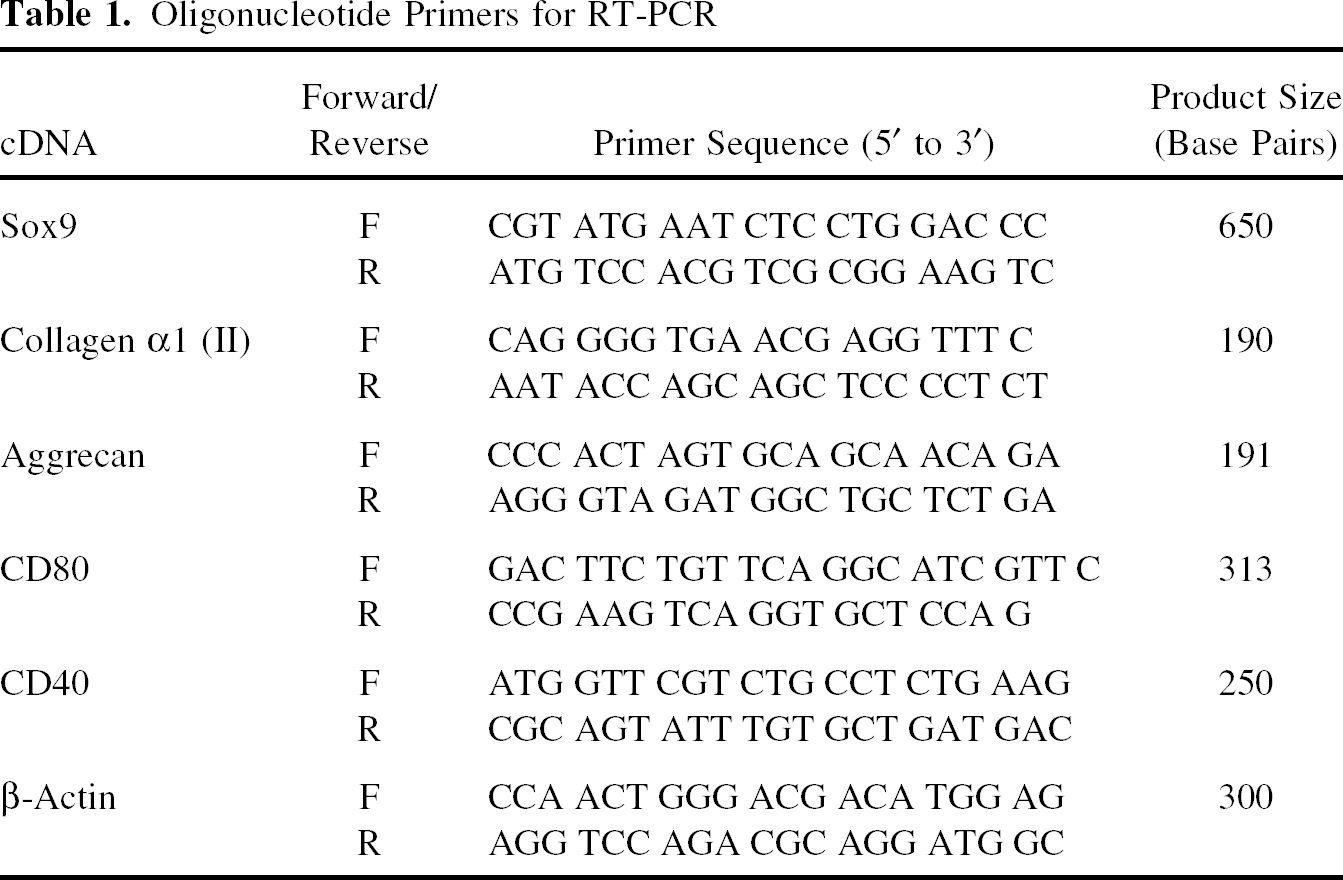
Oligonucleotide Primers for RT-PCR
Flow Cytometric Expression Analysis
Cultured PCC, either untreated or after 24-h incubation with the indicated human cytokines, were harvested, washed, and stained with antibodies to monitor cell surface expression of SLA-I, SLA-II, pig E-selectin (pE-selectin, pCD62E), vascular cell adhesion molecule-1 (pVCAM-1, pCD106), intracellular adhesion molecule-1 (pICAM-1, pCD54), CD86 (pCD86), pCD80, and pCD40. Indirect immunofluorescence of SLA class I and class II was assessed with the specific mouse monoclonal antibodies PT85A and MSA3 (VMRD, Inc., Pullman, WA), respectively. The mAb 1.2B6 (Serotech, Raleigh, NC) was used to stain E-selectin, whereas pVCAM-1 and pICAM-1 were respectively detected with the mouse monoclonal antibodies 3F4 and 3A3.81 (Alexion Pharmaceuticals, Cheshire, CT). Cell surface expression of pCD86 was confirmed with 5B9.88 mAb (Alexion Pharmaceuticals). Specific reactivity to pCD80 and pCD40 was determined with the rabbit polyclonal antibodies ALP61 (Alexion Pharmaceuticals) and H-120 (Santa Cruz Biotechnology, Santa Cruz, CA), respectively. Goat anti-mouse or anti-rabbit IgG fluorescein isothiocyanate (FITC)-conjugated antisera were used as the corresponding secondary antibodies (Invitrogen). Antibody incubations were conducted at 10 μg/ml in PBS/1% FBS for 30 min at 4°C in V-bottom plates. FL-1 fluorescence was measured with a FACScalibur (BD Biosciences).
Flow Cytometric Cell Death Analysis
Confluent PCC or PAEC in 12-well plates were left untreated or preincubated with 10 ng/ml hTNF-α, hIL-1α, or hIL-1β for 24 or 72 h. In some wells, cycloheximide (CHX, Sigma) was added at a final concentration of 2.5 μg/ml. To measure total cell death, all cells were collected from each well, washed, and stained with 2 μg/ml propidium iodide (Sigma) in PBS for flow cytometry. To assess apoptosis, we determined the proportion of hypodiploid (sub-G1) cells after overnight fixation with cold 70% ethanol and staining with 40 μg/ml propidium iodide (Sigma)/100 μg/ml RNasa (Invitrogen) in PBS. Fluorescence was measured with FACScalibur on FL-3.
Static Adhesion Assay
Confluent PCC in 96-well plates were left untreated or preincubated with 10 ng/ml hTNF-α, hIL-1α, or hIL-1β for 24 h as preparation for the adhesion assay. Triplicates for each assay condition were conducted. Immediately before the assay, the plate was washed and kept at room temperature for 30 min with the anti-pVCAM-1 3F4 and the isotype control antibodies in designated wells at 2x concentration (20 μg/ml). The U937 effector cells were simultaneously washed and loaded with Calcein AM (10 μg/ml, Invitrogen) for 20 min at 37°C. All the assay incubations and washes included HBSS (Invitrogen)/1% BSA (Sigma). Calcein-loaded cells were washed and added in equal volume to the PCC in triplicates at 1:1 and 5:1 effector/target (E/T) ratios. After a 45-min incubation at 37°C, nonadherent cells were removed with thorough washes and the remaining cells were lysed with 1% SDS for fluorescence intensity measures with a cytofluorometer (FLUOstar OPTIMA, BMG Labtechnologies, Offenburg, Germany).
PCC and U937 Coculture Experiments
Confluent PCC in 96-well plates were left untreated or preincubated with the indicated cytokines. PCC were subsequently washed with PBS and cocultured with U937 at 1:1 and 5:1 E/T ratios. Each assay condition was conducted in duplicate. After 8 and 24 h of coculture, cell-free supernatants were collected and assayed for hIL-8 by enzyme amplified sensitivity immunoassay (EASIA, Invitrogen) and porcine IL-8 (pIL-8) by ELISA (R&D Systems, Minneapolis, MN). Identical coculture experiments were performed to determine pIL-6 in supernatnats by ELISA (R&D Systems). Determinations were done on a microplate reader (Biotek Instruments, Winooski, VT) at 450 nm.
Costimulation Assay
PCC were seeded to confluency in 96-well plates 24 h before coculturing with Jurkat at 2:1 and 5:1 E/T ratios. Cocultures were kept in duplicates for 24 h, 3 days, or 5 days, as indicated. In experiments determining the contribution of pCD86 and pVCAM-1, selected wells in triplicate were preincubated with isotype-matched control, anti-pCD86 (5B9.88) or anti-pVCAM-1 (3F4) blocking mAb for 30 min prior to (2x) and during the 24-h coculture (5 μg/ml). The phytohemagglutinin (PHA, Sigma) was added to a final concentration of 10 μg/ml at the time of initiating the cocultures and incubated at 37°C for the indicated times and conditions. All experiments included as positive control Jurkat incubated with PHA and anti-human CD28 (hCD28) CD28.2 (5 μg/ml, BD Biosciences). Cell-free supernatants were collected and assayed for hIL-2 by ELISA (Invitrogen). Determinations were done on a Biotek microplate reader at 450 nm.
Statistical Analysis
The indicated values are expressed as the mean ± SEM. Statistical analysis was carried out using the Student t-test when comparing two groups and ANOVA (applying Tukey testing) for multiple comparisons. Differences were considered statistically significant at p ≤ 0.05.
Results
Isolated Porcine Costal Chondrocytes Express Cartilage-Specific Markers
For this study, we isolated chondrocytes from pig costal cartilage because of technical advantages (easy accessibility and low contamination risk) and its properties to form stable tissue-engineered cartilage (40). To ensure that the PCC tested maintained the characteristic traits of cartilage, we assessed by RT-PCR the expression of pig Sox9, type II collagen, and aggrecan at various passages after isolation and culture (Fig. 1). The β-actin control confirmed the good quality of the cDNA in all samples, whereas no amplification of the fragments of interest was observed with PAEC cDNA or in the water control. Chondrocytes analyzed at passages 3 to 6 showed expression of all three cartilage-specific markers (Fig. 1). By contrast, aggrecan expression seemed reduced in PCC at passage 7 and no Sox9 or type II collagen were detected at this stage under the conditions assayed. Moreover, PCC at higher passages than passage 7 showed little or no expression of these markers (data not shown), indicating they were dedifferentiating. Therefore, subsequent experiments were carried out with PCC at passages 3 to 6.
Characterization of PCC by RT-PCR. Expression of pig Sox9, type II collagen, and aggrecan was assessed by RT-PCR after culture of PCC at various passages. A β-actin control for quantity and quality of the cDNA was included. The corresponding samples were loaded for electrophoresis in the following order: lane 1, PCC at passage 3; lane 2, PCC at passage 5; lane 3, PCC at passage 6; lane 4, PCC at passage 7; lane 5, PAEC control; lane 6, water control.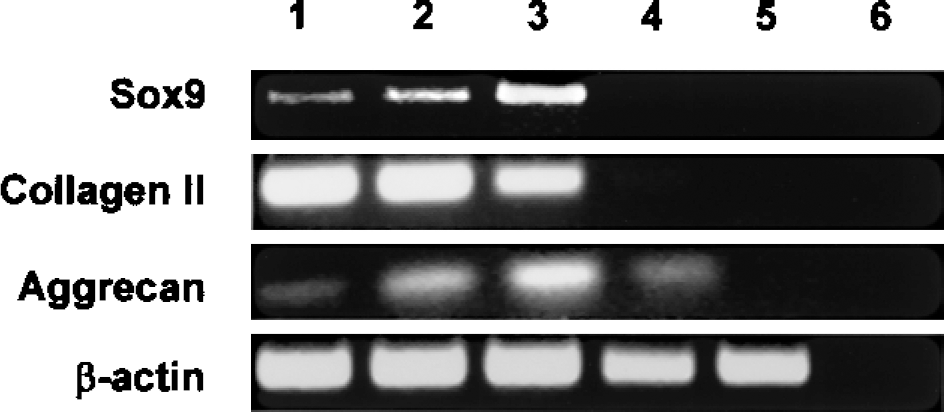
Resting and Cytokine-Stimulated PCC Express SLA I, Adhesion, and Costimulatory Molecules
To understand the PCC capabilities to directly trigger a cellular immune response, we first studied the cell-surface expression of the major histocompatibility complex, adhesion and costimulatory molecules by flow cytometry (Fig. 2). SLA class I, but not SLA II, was detected in resting and cytokine-stimulated conditions (Fig. 2A, B). We also confirmed constitutive and induced expression of pVCAM-1 and pICAM-1 on PCC (Fig. 2A, B), whereas pE-selectin remained undetectable under the multiple conditions assayed (Fig. 2A). In fact, pVCAM-1 was expressed at low levels in unstimulated PCC, but reached very high levels after cytokine treatment. In contrast, the potent costimulatory molecule pCD86 was expressed at moderate levels in both resting chondrocytes and after stimulation with hTNF-α and hIL-1 cytokines (Fig. 2A, B). Finally, pCD80 and pCD40 were barely detected on the cell surface regardless of the treatment (Fig. 2A). We ruled out any lack of function of the reagents utilized for our negative determinations, as all reacted positively to the appropriate controls (data not shown). Nevertheless, we decided to confirm specific expression by RT-PCR of these two costimulatory molecules because of their functional relevance (Fig. 2C). We amplified specific PCR products for both pCD80 and pCD40 in untreated PCC (Fig. 2C).
Expression of SLA, adhesion, and costimulatory molecules on resting and cytokine-stimulated PCC. PCC, either untreated or after 24-h incubation with the indicated human cytokines, were monitored for cell-surface expression of immunoregulatory molecules. (A) Specific staining for SLA I and II, pVCAM-1 and pICAM-1, pE-Selectin, pCD86, pCD80, and pCD40 was determined on resting PCC by flow cytometric analysis. The dotted or dashed line in each panel corresponds to the secondary antibody control (2Ab). (B) The effect of hTNF-α, hIL-1α, and hIL-1β on cell-surface expression of SLA I, pVCAM-1, pICAM-1, and pCD86 is depicted as the mean ± SEM of the mean FL-1 fluorescence intensity corresponding to five experiments. Significant differences were observed between untreated and cytokine-stimulated PCC for each molecule when indicated: *p < 0.05, **p < 0.001. (C) Expression of pCD80 and pCD40 was assessed by RT-PCR after culture of PCC at various passages. A β-actin control for quantity and quality of the cDNA was included. The corresponding samples were loaded for electrophoresis in the following order: lane 1, PCC at passage 5; lane 2, PCC at passage 6; lane 3, PAEC control; lane 4, pig monocytes control; lane 5, CHO control; lane 6, water control. nd, not determined.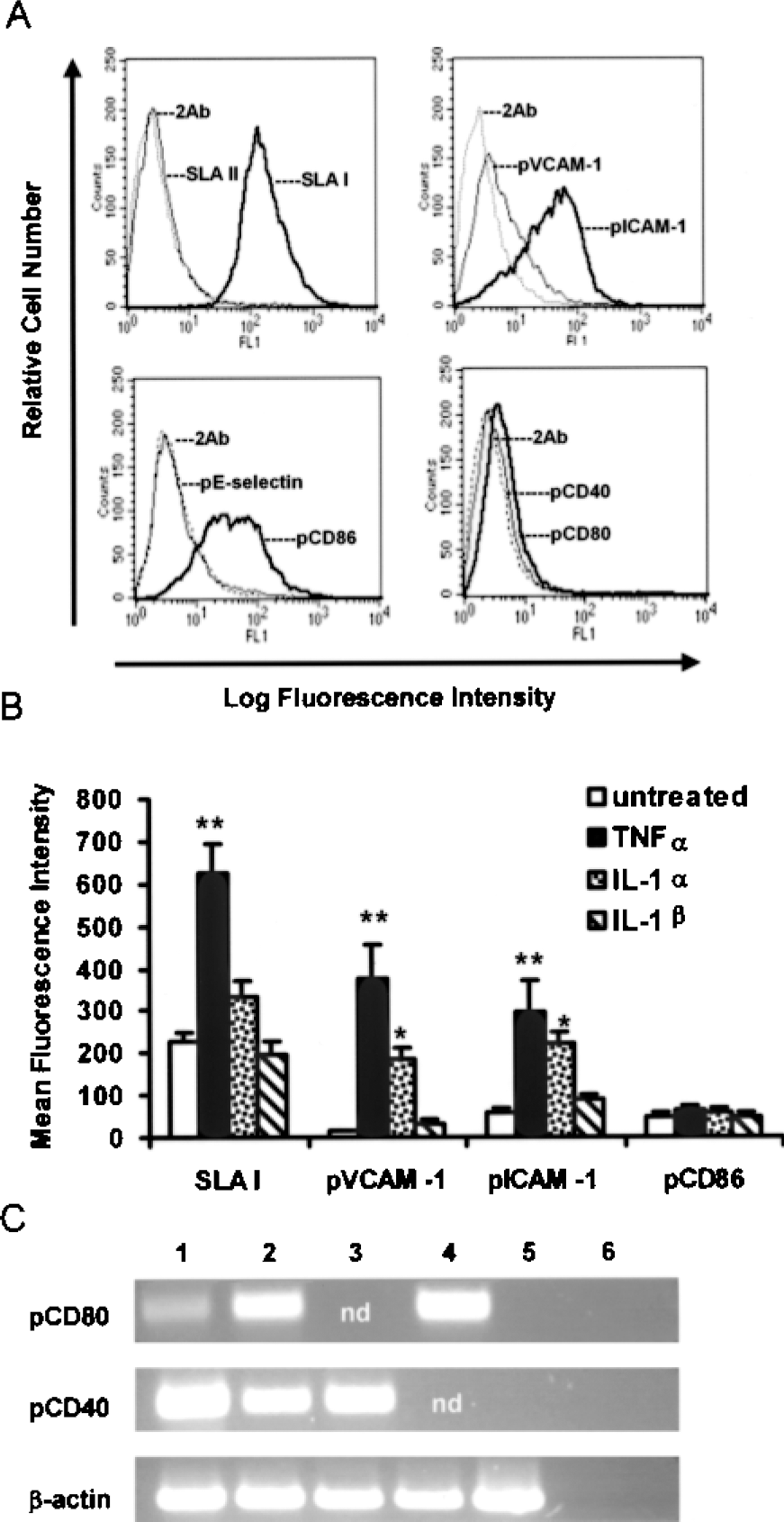
To study the regulation of these molecules by proinflammatory cytokines, we chose to use hTNF-α, hIL-1α, and hIL-1β for its relevance in cartilage inflammation and its potential role in xenograft rejection. The hTNF-α produced the most potent effect leading to a dramatic and significant upregulation of SLA I, pVCAM-1, and pICAM-1 (Fig. 2B). The hIL-1α and to a lesser extent hIL-1β enhanced expression of the adhesion molecules pVCAM-1 and pICAM-1. This particular effect of the hIL-1 cytokines was highly reproducible, despite it only reached significance for hIL-1α due to variability between assays. On the contrary, hIL-1α had a minor effect on SLA I expression and hIL-1β simply did not upregulate SLA I (Fig. 2B).
PCC Are Highly Resistant to Cytokine-Mediated Cell Death
To better understand the effect of hTNF-α, hIL-1α, and hIL-1β on PCC, we also assessed whether these cytokines could induce apoptosis of PCC and compared it to that of PAEC. As a measure of apoptosis, we determined the percentage of hypodiploid cells after incubation with cytokines for 24 and 72 h in the presence or absence of CHX (Fig. 3). In parallel studies, we measured total cell death and observed comparable results (data not shown). None of the cytokines, when tested without CHX, induced apoptosis of chondrocytes above background even after incubation for 72 h (Fig. 3A). Under identical conditions, hTNF-α induced a certain degree of apoptosis in PAEC that increased over time (26.6 ± 7.4% vs. 2.9 ± 0.4% for untreated control at 72 h). Addition of CHX to the various treatments increased the percentage of apoptotic PCC overall and specifically in response to hTNF-α (Fig. 3B). This is better appreciated at 24 h (25.3 ± 7.5% vs. 13.6 ± 3.1% CHX control), as more cells succumb to CHX later on. However, we did not observe significant differences for PCC between the various conditions with exception of the CHX effect. Moreover, higher concentrations of CHX did not further enhance the hTNF-α effect (data not shown). Thus, PCC were less susceptible to cytokine-mediated cell death than PAEC, as most PAEC were dead by 24 h after treatment with hTNF-α and CHX (77.3 ± 5%). CHX alone produced 39.7 ± 7.4% apoptotic PAEC at this time point. To confirm the specificity of the hTNF-α treatment and assess a blocking reagent for this pathway, we also included a combination treatment with pTNFR2Ig (9). This reagent nicely prevented the hTNF-α-mediated apoptosis of PCC (Fig. 3C) and PAEC (data not shown).
Apoptosis of PCC induced by cytokines. Apoptosis of PCC was assessed by flow cytometry by determining the proportion of hypodiploid cells after treatment for 24 and 72 h with hTNF-α, hIL-1α, and hIL-1β in the absence (A) or presence (B) of CXH. The mean ± SEM of four independent experiments is shown. We did not observe significant differences between conditions for PCC with exception of the effect of CHX relative to untreated controls. The hypodiploid PCC population was also measured after adding the blocking reagent pTNFR2Ig to the hTNF-α treatments (C). The results of a representative experiment of four at the 24-h treatment are shown.
Human Monoblastic Cells Adhere to PCC Through pVCAM-1
We next studied the interaction of the human monoblastic cell line U937 with untreated and cytokine-activated PCC in a static adhesion assay testing various E/T ratios (Fig. 4). As previously observed (7), U937 demonstrated significant adherence to PCC. The fluorescence intensity, which determined adhesion, was overall five times higher at 5:1 than at 1:1 E/T ratio on any given assay (data not shown due to data normalization). The effect of cytokines was best demonstrated at the 1: 1 E/T ratio in which hTNF-α increased adhesion fourfold and hIL-1α twofold, whereas hIL-1α had little effect (Fig. 4A). At the 5:1 ratio these elevations were down by half (2- and 1.5-fold, respectively) (Fig. 4B). We took advantage of the blocking activity of the anti-pVCAM-1 mAb 3F4 and tested whether pVCAM-1 contributed to the adherence of PCC by U937 cells. U937 adhesion to PCC at 1:1 E/T ratio was not affected by 3F4 under resting conditions, but it was markedly reduced to almost basal levels for PCC pretreated with hTNF-α or hIL-1α (Fig. 4A). An isotype-matched IgG1 control mAb or the anti-pCD86 5B9.88 mAb (also an IgG1) had no such effect (both showed comparable results). At 5:1 E/T ratio, the effect of pVCAM-1 in untreated or hIL-1α-treated PCC was slightly more apparent (although not significant), but it was unexpectedly overwhelmed after activation with hTNF-α (Fig. 4B). In a few experiments, we added a blocking antibody for human CD49d, either alone or in combination with 3F4, but it did not provide further reduction in adherence (data not shown).
Static adhesion of human monoblastic U937 cells to PCC. The adherence of calceinloaded U937 to confluent PCC was measured as described in Materials and Methods. PCC were left untreated or preincubated with 10 ng/ml hTNF-α, hIL-1α, or hIL-1β for 24 h prior to the assay. The anti-pVCAM-1 mAb and the isotype-matched control antibodies were added as indicated. Here we present the mean ± SEM of four independent experiments at 1:1 (A) and 5:1 (B) E/T ratios after normalizing relative to basal adhesion. For statistical purposes, results of the irrelevant isotype-matched control and the anti-pCD86 mAb were pooled together (1 of 4 at 1:1 and 2 of 4 at 5:1 E/T ratios). Significant differences were observed for the effect of cytokines when comparing conditions with isotype control mAb (#p < 0.05, ##p < 0.001) and for the effect of anti-pVCAM-1 relative to the isotype control mAb and media control within the same treatment (*p < 0.05).
PCC Secrete pIL-8 and pIL-6 in Response to Human Cytokines and Induce U937 to Secrete hIL-8
IL-8 Concentrations in Supernatants of U937 and PCC Cocultures

Cocultures of U937 effector cells (E) and PCC, the target cells (T), were kept for 8 and 24 h. The PCC were untreated or pretreated with the indicated cytokines for 24 h. Representative experiment of 3. nd, not determined.
The hIL-8 was determined in the same harvested supernatants to assess the U937 response (Table 2). We encountered the limitation that the EASIA kit utilized for hIL-8 determinations cross-reacted partially with the pig homologue, producing some positive reactivity in supernatants of hTNF-α-pretreated PCC not cocultured with U937. Nevertheless, we detected little or no reactivity for the rest of the PCC cultured alone. Moreover, the hIL-8 increased (as opposed to pIL-8) in correlation with the E/T ratio and in response to the pretreatment with cytokines. Thus, these increases in hIL-8 secretion were more pronounced after coculture with cytokine-activated PCC.
PCC Activate Jurkat T Cells Through pCD86
To assess whether PCC could provide costimulatory signals to human T cells, we cocultured PCC with Jurkat in the presence of PHA and determined hIL-2 secretion in culture supernatants. In the first set of experiments, we measured hIL-2 release after 1, 3, and 5 days of co-culture at 2:1 and 5:1 E/T ratios (Fig. 5). In the absence of a primary signal (no PHA), Jurkat (either alone or cocultured with PCC) did not produce hIL-2 (Fig. 5A, B). Addition of PHA to Jurkat alone led to hIL-2 secretion in very small amounts, almost undetectable at 2:1 E/T ratio. On the contrary, coculture with PCC provided a very strong costimulatory effect in the presence of PHA at both E/T ratios (Fig. 5A, B). Moreover, addition of the anti-hCD28 agonist mAb to the coculture did not provide further stimulation over time, whereas Jurkat alone plus PHA and anti-hCD28 showed equal (to 5:1 ratio; Fig. 5B) or less (to 2:1 ratio; Fig. 5A) activation. Saturation of the system was certainly reached at 5:1 ratio at an early time point (Fig. 5B), whereas the maximum hIL-2 levels were observed between 3 and 5 days of the 2:1 E/T ratio coculture, depending on the experiment (Fig. 5A).
Coculture experiments to determine Jurkat activation by PCC through costimulatory signals. Jurkat were left untreated or activated with PHA or PHA plus anti-hCD28 (aCD28) in the absence or presence of PCC and kept in culture for 1, 3, and 5 days. Coculture experiments were conducted at 2:1 (A) and 5:1 (B) E/T ratios. Jurkat activation was assessed by determining hIL-2 concentration in culture supernatants by ELISA. These results are representative of three independent experiments.
Taking into account these results, we chose to coculture Jurkat and PCC for 24 h at 2:1 E/T ratio to assess the participation of pCD86 and pVCAM-1 in this process (Fig. 6). In the presence of PHA, pCD86 blockade markedly reduced hIL-2 secretion by 80–90% compared to control cocultures in all assays. Blockade of pVCAM-1 also diminished hIL-2 secretion, but differences relative to controls were less pronounced and with higher variability between experiments (10–50% reduction). Moreover, the combination of the two blocking antibodies led to little further reduction (data not shown), consistent with pVCAM-1 playing a role secondary to pCD86 in T-cell costimulation mediated by PCC.
Coculture experiments to determine the contribution of pig CD86 and VCAM-1 to PCC-mediated Jurkat activation. Jurkat were left untreated or activated with PHA or PHA plus anti-hCD28 in the absence or presence of PCC at 2:1 E/T ratio. When indicated, PCC were pretreated with anti-pCD86, anti-pVCAM-1, or isotype-matched control mAb. Jurkat activation was assessed by determining hIL-2 concentration after 24 h in culture supernatants by ELISA. A representative experiment of five independent experiments is shown. Statistical differences were observed between the means of triplicates corresponding to the anti-pCD86 and the anti-pVCAM-1 mAb treatments versus isotype control mAb. *p < 0.05, **p < 0.01.
Discussion
In this work, we studied the molecules and systems involved in the development of a cellular immune response to xenogeneic pig chondrocytes. PCC responded to various human cytokines and interacted with human immune cells through adhesion and costimulatory molecules. In particular, we identified the cytokines TNF-α and IL-1α and the pig membrane-bound proteins CD86 and VCAM-1 as potential targets for intervention in cartilage/chondrocyte xenotransplantation. This study has special relevance because of the feasibility to engineer the donor pig cell to counteract some or all of these molecules to promote engraftment. Previous work identified the Gal antigen as a key trigger of the humoral and cellular response against pig cartilage that could be averted by genetic manipulation (7,8). The incorporation of additional modifications that target the cellular response may help to overcome rejection of the xenogeneic cartilage.
We characterized our PCC monolayer cultures for expression of cartilage markers and worked with chondrocytes that expressed Sox9, type II collagen, and aggrecan for these studies. The transcription factor Sox9 is key for chondrocyte differentiation and enhances expression of type II collagen and other cartilage-specific genes (22). Accordingly, expression of type II collagen in monolayer cultures of adult human articular chondrocytes has been shown to correlate well with their capacity to produce hyaline-like cartilage in vivo (13). In support of our work in the xenotransplantation field, Fuentes-Boquete et al. have demonstrated that pig chondrocytes have no impediments in producing good quality cartilage to repair a human cartilage defect in vitro (in the absence of an immune response) (16).
As a first step towards identifying key molecules and pathways that participate in the cellular rejection process, we determined the presence or absence of a good number of immunoregulatory molecules in PCC in resting and stimulated conditions. In both circumstances, we observed expression of SLA I but not SLA II on PCC, which implies that PCC may directly activate CD8+ T cells through the TCR, but do not have the ability to present antigen to CD4+ T cells. The lack of SLA II detection after the cytokine treatments was unexpected as MHC class II is expressed in human and other mammalian chondrocytes (24) and in PAEC activated with hTNF-α (2,6). Nevertheless, these results are consistent with previous observations in pig arthritic joints in which the majority of chondrocytes expressed high levels of ICAM-1 but no SLA II (11). Moreover, SLA II cannot be upregulatated by human IFN-γ because the porcine receptor does not respond to human IFN-γ (34). Regarding the expression of adhesion molecules, we detected pVCAM-1 and pICAM-1 on PCC and both were strongly induced by hTNF-α and hIL-1α. Their expression profiles are actually very similar to those reported for human articular chondrocytes (20). In addition, we determined the absence of E-selectin on pig chondrocytes under the various conditions assayed, which indicates that pE-selectin does not contribute to pig cartilage rejection. Finally, we detected constitutive expression of pCD86 on PCC, but little pCD80 and pCD40. The functional implications of this expression pattern for adhesion and costimulatory molecules are discussed below.
Human TNF-α and IL-1α were confirmed as potent inducers of a proinflammatory phenotype in chondrocytes, whereas hIL-1β had less effect. The fact that these cytokines showed little capacity to induce cell death of PCC suggests this is not a relevant mechanism in their contribution to the rejection process. Moreover, these findings discard that these cytokines induce an apoptotic phenotype that causes the increased adhesion to human monocytes. On the contrary, our expression analyses indicate that hTNF-α promotes both adhesion and antigen presentation by direct upregulation of key cell-surface molecules, whereas the hIL-1 cytokines primarily enhance adhesion following a similar mechanism. Accordingly, adherence of the human monoblastic cells to PCC paralleled well the upregulation of pVCAM-1 and pICAM-1 in response to each of these cytokines. The relevant contribution of pVCAM-1 after cytokine stimulation was further confirmed by the marked reduction in adhesion observed at a low E/T ratio when using a specific blocking mAb. From these experiments, we can conclude that pVCAM-1 (by binding human CD49d) is responsible for the increased adhesion observed after treatment with hIL-1 cytokines and it participates in adhesion to hTNF-α-stimulated PCC. We have previously shown that the Gal antigen promotes U937 adhesion to porcine chondrocytes either unstimulated or after treatment with hTNF-α (7). Therefore, we expect the Gal epitope to be mainly contributing to basal adhesion. However, other molecules, most probably ICAM-1 (which binding is preserved cross-species) (39), must be playing a role in this process, especially after hTNF-α treatment. We also speculate that conformational changes in integrins may be counteracting the effect of the VCAM-1 blockade on U937 adhesion to hTNF-α-stimulated PCC at a high E/T ratio. Overall, our results indicate that PCC bind U937 through at least pVCAM-1 and the Gal antigen. These ligands are known to participate in the human monocyte–PAEC interactions (19,21).
In the current study, we have elucidated the biological activity of hTNF and hIL-1 cytokines on pig chondrocytes at multiple levels. Whereas our previous work pointed out a potential enhancement of human monocyte adhesion to pig articular chondrocytes by hTNF-α (7), we have now confirmed this effect on costal chondrocytes, identified molecules that may be involved in this process and compared it to that of hIL-1 cytokines. We have also revealed their effect on apoptosis and total cell death of PCC. Finally, these human cytokines also stimulated secretion by PCC of pIL-6 and the potent chemokine pIL-8. As with cell-surface molecules, hTNF-α produced a greater release of these factors than the hIL-1 cytokines by 24 h. Although this is the first report of pIL-8 and pIL-6 secretion by pig chondrocytes, induced expression of pig IL-1α and IL-1β mRNA was detected in a model of pig cartilage explantation and cutting (17). Moreover, numerous studies describe how human chondrocytes secrete multiple cytokines and chemokines during arthritis and in response to cytokines, a process that is at least partially mediated by NF-κB (12,27). It is highly likely that PCC themselves are capable of inducing a proinflammatory response in a human setting, as many cytokines and chemokines function crossspecies. In fact, we observed that the amount of pIL-8 inversely correlated with the number of U937 in the cocultures. Interestingly, we also detected increased hIL-8 release after U937 and PCC cocultures, suggesting that cytokine-activated PCC may stimulate the human leukocytes to produce inflammatory mediators. Similarities can be found with the induced activation of human DC after direct contact with PAEC (23). Thus, both human monocytes and pig chondrocytes may contribute to the inflammatory milieu of xenograft rejection.
Human T cells also have the machinery to recognize PCC and produce an immune response, as Jurkat T cells cocultured with PCC released high amounts of hIL-2 in the presence of PHA. This kind of assay reproduces well the response of highly purified T cells to PAEC and is considered appropriate to evaluate the effect of costimulatory molecules (3,26,28). Thus, we can conclude from our experiments that pCD86 on PCC provides a key costimulatory signal for human T cells, whereas pVCAM-1 plays a secondary role. No synergism was observed between the pCD86 and pVCAM-1 pathways. Other costimulatory molecules such as CD58 may be responsible for the remaining Jurkat stimulation and will be tested as well when we obtain the appropriate reagents. It is unclear at this point whether the poorly expressed pCD80 and/or pCD40 can contribute significantly to the T cell response. For comparison, PAEC also express pCD86, which plays a major role mediating activation of human T cells (26). However, contrary to PCC, PAEC express pCD40 at readily detectable levels on the cell surface (29). The studies of pCD40 have focused on its effect on PAEC, but pCD40 may well participate in the activation of human T cells as an anti-hCD154 mAb displays an inhibitory effect in coculture experiments (35).
The blocking antibodies used for these experiments, the anti-pVCAM-1 3F4 and the anti-pCD86 5B9.88, have been successfully used in the past when applied to other xenogeneic settings such as with pig endothelial cells cocultured with human monocytes, NK cells and Jurkat T cells (18,21,26). Their specificity has been well established by us and other researchers (5,18,21,26). We are aware that there is a risk for unspecific binding through the Fc receptors when using blocking antibodies in our system. However, these are mouse IgG1 antibodies (that bind less than human antibodies to human Fc receptors) and we included in all cases isotype-matched mAbs (even mAbs that we knew would bind to the PCC cell-surface such as the anti-pCD86 5B9.88). We did not observe any significant alterations in adhesion due to the use of the control antibodies that would lead to question our conclusions regarding the reduction in adhesion observed with the anti-pVCAM-1 blocking antibody, especially at the 1:1 E/T ratio that produced the most clear and robust results. If there were effects expected from the binding of Fc regions and/or recognition of any xenogeneic elements in the antibodies such as carbohydrates, which would lead to an increase not a reduction in adhesion. In fact, we actually observed a slight increase in adhesion that did not reach significance at the 5:1 E/T ratio. Likewise, the consistently distinct effect of the four different antibodies used for the costimulatory experiments with Jurkat T cells, including the positive and negative controls, further confirmed their specificity. Finally, it is also true that chondrocytes are difficult to genetically modify in vitro, more than many other primary cultured cells. Thus, the use of antibodies seems appropriate using the right controls compared to other approaches that would be much more technologically challenging. The siRNA technology is certainly another interesting tool to assess the role of specific molecules. Although we did not consider using siRNA for these experiments, they may be useful to reach similar conclusions to those obtained with the antibodies and to test the contribution of other molecules in future studies.
In summary, we have identified various cell-surface molecules and cytokines that contribute to the xenogeneic cellular immune response triggered by pig chondrocytes. Although there are clear parallelisms with the mechanisms described for PAEC, there are differences as well. The lack or low expression of some key molecules such as SLA II, pCD40, and pE-selectin suggests that PCC may induce a milder cellular immune response than porcine endothelial cells. Regarding susceptibility to cytokine-induced apoptosis, we also observed a higher resistance in the case of PCC. In addition, cartilage is known to have some immune privilege properties that need further clarification (24). Thus, these particularities of pig chondrocytes may reflect differences between rejection of cell-based/avascular tissue and solid organ xenografts. Our findings further support the concept that there are lesser barriers to attain engraftment for cellular xenografts. Therefore, it may be easier to modify the donor cell to the point of allowing long-term graft survival. Because of their strong activity, TNF and pCD86 are primary targets for intervention for the development of clinical applications based on xenotransplantation of pig chondrocytes/cartilage. Here, we already show how some effects of their specific blockade may help avert the cellular immune response against pig chondrocytes. Interestingly, we have previously developed approaches based on genetic engineering of the donor cell that could be applied to counteract TNF and pCD86 (5,6,9,10). Thus, our results encourage us to continue studying the molecular bases of xenogeneic cartilage rejection and developing the strategies that promote pig chondrocyte engraftment for future clinical applications.
Footnotes
Acknowledgments
We gratefully acknowledge A. Gimeno for helping in the porcine tissue harvest and M. Uribe-Herranz with the isolation of porcine chondrocytes. This work was mainly supported by a grant from Fundación de Investigación Médica Mútua Madrileña (338/05) and partially supported by Ministerio de Educación y Ciencia (SAF2005-00472), both to C.C. C.C. was also supported by the Ramón y Cajal program from Ministerio de Educación y Ciencia (Spain), an aid for emerging groups from Generalitat de Catalunya (2005SGR00897), and a Marie Curie action from the European Commission (MIRG-CT-2005-021293).