
Abstract
Duchenne muscular dystrophy is caused by the absence of functional dystrophin, leading to the myofiber membrane instability and progressive muscle atrophy. Myoblast transplantation in dystrophic muscles is a potential therapy, as it permits the long-term restoration of dystrophin expression in transplanted muscles. However, the success of this approach is limited by the short period of muscle repair following myoblast transplantation. Myostatin, a powerful inhibitor of muscle growth, is involved in terminating the period of muscle repair following injury by reducing myoblast proliferation and differentiation. Follistatin forms a complex with myostatin, preventing its interaction with its receptor and thus blocking the myostatin signal. Here, we used a lentivirus to overexpress the follistatin protein in normal myoblasts to block the myostatin signaling. We measured the potential of transduced myoblasts to proliferate and to form multinucleated myotubes in vitro. And finally, we considered the engraftment success of those transduced myoblasts in comparison with control cells in vivo within SCID mice TA muscle. Our results first confirmed the overexpression of follistatin into lentivirus transduced myoblasts, and second showed that the overexpression of the follistatin in normal human myoblasts improved in vitro their proliferation rate by about 1.5-fold after 96 h and also their differentiation rate by about 1.6- and 1.8-fold, respectively, in the absence and in the presence of recombinant myostatin. Finally, our data demonstrated that the engraftment of human normal myoblasts overexpressing the follistatin protein into SCID mouse muscles was enhanced by twofold.
Introduction
Duchenne muscular dystrophy (DMD), a severe X-linked genetic disease, is the most severe dystrophy in children. It affects one male in 3,500 at birth (3). The disease is due to a mutation of the dystrophin gene, resulting into the absence of that submembrane protein. Dystrophin is a 427-kDa protein expressed in myofibers, where it provides a link between the extracellular matrix and the cytoskeleton (6). The absence of dystrophin induces an instability of the myofiber membrane and progressive degeneration of muscle tissue (25). Normal myoblasts transplanted in dystrophic muscles fuse with damaged host myofibers, forming hybrid fibers, which express dystrophin (21). The limited period of muscle regeneration that follows muscle injury reduces the success of the therapeutic approach. This can be bypassed by repetitive cell injection very close to each other, as demonstrated by our group (27).
Myostatin, also called growth and differentiation factor-8, belongs to the superfamily of transforming growth factor-β. It is predominantly expressed in skeletal muscle, which suggests that it plays an important role in regulating muscle development (17). The great muscle hypertrophy phenotype of mice lacking myostatin gene suggested that myostatin normally regulates negatively muscle growth (17). In fact, myostatin reduces the proliferation and differentiation of both myoblasts and satellites cells (16, 24, 30). As the other members of the TGF-β family, myostatin is synthesized as a precursor protein that undergoes two proteolytic processing to generate the biologically active molecule (17). The first cleavage removes the N-terminal signal necessary for targeting the protein to the secretion pathway, and the second one generates the C-terminal fragment, which possesses the receptor binding activity. The N-terminal fragment following the second proteolytic processing has been referred as the propeptide; this propeptide plays an important role in regulating the activity of mature myostatin in vivo (13). In addition to the propeptide, several other proteins have also been shown to be capable of binding and inhibiting myostatin (4, 5, 20, 31). One of these is follistatin protein, and this inhibition occurs after the formation of a latent complex formed by active myostatin and follistatin (13). Indeed, it has been reported that transgenic mice overexpressing the follistatin protein show an increased muscle mass similar to that observed in transgenic mice lacking myostatin (2, 13), while a lack of functional follistatin causes a important reduction in muscle mass at birth (15).
In addition, Minetti et al. showed that treatment of mdx mice with deacetylase inhibitor (trichostatin A) results in an increase in the size of myofibers by inducing expression of the myostatin antagonist follistatin in satellite cells, and that this conferred to dystrophic muscles resistance to contraction-coupled degeneration and alleviated both morphological and functional consequences of the primary genetic defect (19). Follistatin is a secreted protein glycoprotein encoded by a single gene that is subject to alternative splicing. This mechanism generates two different forms of the protein: the short (FS288) and the long (FS 315) form of follistatin (26). The short form of follistatin lacks the acidic tail in the C-terminal, which permits the protein to bind the extracellular matrix and thus to be less diluted in the circulation (8).
The objective of our experiments was first to genetically modify human myoblasts with a recombinant lentivirus to overexpress the follistatin short form protein to increase their proliferation and/or differentiation rates and to measure the success of the transplantation of these genetically modified myoblasts in immunodeficient SCID mouse muscles.
Materials and Methods
Animals
All the experiments were approved by the animal care committee of the CHUL (Centre Hospitalier de l'Université Laval). Severe combined immunodeficiency disease (SCID) mice were purchased from Charles River (Toronto, Ontario, CA).
Cell Lines
293T cells were grown in DMEM (Gibco, Burlington, Ontario, CA) proliferation medium complemented with 10% FBS (Invitrogen, Burlington, Ontario, CA) and 1% penicillin-streptomycin (Gibco). Myogenic cells were released from minced fragments of muscle biopsy by enzymatic dissociation. After 1 h of incubation in collagenase (600 U/ml) (Sigma-Aldrich, St. Louis, MO, USA), cells were then incubated for 30 min in Hank's buffer salt solution (HBSS) (Gibco) containing 0.1% trypsin and 0.02% EDTA (Gibco). Dissociated cells were grown in a modified MB 1 medium (Hyclone, South Logan, UT, USA) containing 10% fetal bovine serum (FBS) (Gibco, Burlington, Ontario, CA), 10 ng/ml basic fibroblast growth factor (bFGF) (Strathmann Biotec AG, Hamburg, Germany), and 1% penicillin-streptomycin in a humidified atmosphere with 5% CO2 at 37°C. Differentiation was induced by rinsing the cultures and switching them to DMEM medium containing 2% FBS for 2 days.
Construction of a Lentivirus Vector and Viral Preparation
The follistatin lentivirus vector was constructed in our laboratory from a plasmid coding for the human follistatin short form received from Dr. Lee's laboratory. For that, the follistatin transgene was cloned in a lentivirus vector under the control of a CMV promoter (pCMV-hFst). A lentivirus vector coding for the eGFP was used as a control (Fig. 1a). The day before the transfection, 4 × 106 of 293T cells were plated in 10-cm culture dishes in DMEM proliferation medium supplemented with 10% FBS and 1% penicillin-streptomycin. Cells were then transfected with 15 μg of pCMV-hFst vector using the CaCl2 transfection method to produce the follistatin lentivirus. As a control, 293T cells were transfected with 15 μg of pCMV-eGFP vector. In both cases, cells were cotransfected with 6.5 μg of GP, 3.5 μg of VSVG, and 2.5 μg of REV plasmids to permit the production of the viral particles. Cells were incubated overnight in proliferation medium. The day after transfection, cells were washed with HBSS and fresh proliferation medium was added to each dish. The supernatant containing produced hFst lentivirus and control eGFP lentiviruses were collected from transfected 293T cells 48, 72, and 96 h after transfection. The lentivirus was then frozen immediately in liquid nitrogen and stored at −80°C until transduction.

Overexpression of follistatin in 293T cells transfected with human follistatin lentivirus vector. (a) The human follistatin lentivirus vector was constructed by replacing the eGFP sequence in the control vector by the sequence of the short form of the human follistatin obtained from an expression vector. (b, top) 293T cells expressing the eGFP after the transfection and before the fixation (50×). (b, middle and bottom) Immunocytochemical detection of human follistatin in 293T cells transfected with pCMV-eGFP or pCMV-hFst and corresponding nuclei stained with DAPI (50×). (c) Western blot detection of follistatin in extracts of 293T cells transfected with either the eFGP or the follistatin vector.
Transduction of Normal Human Myoblasts with GFP or hFst Lentivirus
The day before the transduction, 250,000 human myoblasts were plated into six-well plates. Cells were transduced with pCMV-eGFP or pCMV-hFst lentiviral particles in the presence of 6 μg/ml of polybrene. Cells were washed twice with HBSS 24 h after transduction to remove all viral particles. Cells were then proliferated. During the culture, cells were transferred in T-75 flasks.
Detection of Human Follistatin by Immunocytochemistry
The eGFP was directly visualized under a fluorescent microscope. An immunocytochemistry assay was performed on pCMV-eGFP and pCMV-hFst lentivirus transduced cells to verify the expression of the human follistatin in transduced cells. Cells were first fixed for 15 min with ethanol 95% and nonspecific binding sites were blocked for 1 h with PBS containing 10% FBS. Cells were first incubated with monoclonal mouse anti-human follistatin primary antibody (R&D Systems, Minneapolis, MN, USA) in PBS 1% FBS (at 25 μg/ml) for 1 h followed by an anti-mouse IgG conjugated with Alexa 546 (Molecular Probes, Burlington, Ontario, CA) (1:300) for 1 h. Finally, nuclei were stained with DAPI.
Detection of the Human Follistatin by Western Blot
A Western blot was made with extracts from pCMV-eGFP and pCMV-hFst lentivirus transduced cells to quantify the expression of human follistatin. Cells were first sonicated directly in a lysis buffer containing 20 mM Tris, pH 7.5, 1 mM DTT, 1 mM PMSF, 1% SDS. Proteins were then precipitated with methanol and chloroform and finally quantified using the amino-black reagent. After that, 20 μg of protein was separated by SDS-PAGE under reducing conditions. Follistatin was detected by incubating the membrane with a monoclonal mouse anti-human follistatin antibody at 25 μg/ml (R&D Systems) and incubating for 1 h. An anti-mouse horseradish peroxidase-coupled antibody (DAKO, Carpinteria, CA, USA) was used as a second antibody at 1: 2000 and incubated for 1 h.
Immunodetection of Human Follistatin Protein by Dot Blot
A dot blot against human follistatin was performed with the conditioned media of human cells transduced with the pCMV-eGFP or with the pCMV-hFst lentivirus to verify the follistatin secretion. Then 100 and 500 μl conditioned medium from eGFP and hFst transduced cells was applied to a nitrocellulose membrane. The membrane was then incubated with a monoclonal mouse anti-human follistatin antibody at 25 μg/ml (R&D Systems) for 1 h followed by an anti-mouse horseradish peroxidase-coupled antibody at 1:2000 for 1 h.
Proliferation Test
pCMV-eGFP and pCMV-hFst lentivirus transduced human myoblasts (2,000 each) were plated in four 24-well plates in proliferation medium (DMEM-HG, 10% FBS, 1% penicillin-streptomycin). Cells were synchronized in serum-free medium for 48 h before the experiment. The proliferation medium was replaced daily. Cells were rinsed with PBS, blotted, and frozen at −80°C until analysis. Proliferation was measured at T0 (48 h after the serum starvation), T48h, T72h, and T96h using the Cyquant cell proliferation kit (Molecular Probes) assay according to the manufacturer's protocol. The assay was performed three times.
Fusion Index
pCMV-eGFP- and pCMV-hFst transduced cells were plated in a 24-well plate at 40,000 cells/well in proliferation medium. One day later, cells were rinsed with HBSS and incubated in a differentiation medium (DMEM containing 2% FBS), in the presence or in the absence of recombinant myostatin in a humidified atmosphere with 5% CO2 at 37°C for 2 days. Fresh differentiation medium with or without recombinant myostatin was replaced daily. Cells were then fixed in 4% paraformaldehyde for 15 min, and permeabilized with 3% Triton X-100 in PBS for 3 × 15 min. Myotubes were immunostained with a mouse anti-myosin heavy chain antibody (1:100 for 2 h), followed by an anti-mouse IgG conjugated with Alexa 546 (1:300 for 1 h). Cell nuclei were marked with DAPI (1:10000). After the immunocytochemistry, the fusion index (defined as the number of DAPI-stained nuclei inside myotubes in a given field divided by the total number of DAPI-stained nuclei in the same field) of each condition was calculated. The assay was performed three times.
In Vivo Animal Studies
hFst transduced human myoblasts (500,000) were transplanted in tibialis anterior muscles of three 3-month-old SCID mice. Control muscles were transplanted with the same amount of cells transduced with the eGFP lentivirus. For transplantation, cells were detached from the flasks using Trypsin-EDTA and washed twice with HBSS. The cell pellet was resuspended in 10 μl of HBSS and slowly injected in muscles using a glass capillary. Mice were sacrificed 3 weeks later and the cell-grafted muscles were dissected out and frozen. Serial cryostat sections (12 μm) were prepared throughout the whole muscle length.
Immunohistochemical Detection of Human Dystrophin
Sections were first washed with PBS. Nonspecific binding sites were blocked by incubating the cryostat sections with PBS containing 10% FBS for 1 h. Human dystrophin was detected with a mouse anti-human dystrophin antibody (Vector Laboratories, Burlington, Ontario, CA) (1:50 for 2 h), followed by a biotinylated anti-mouse antibody (DAKO) (1:300 for 45 min) and streptavidin-Cy3 (Sigma-Aldrich) (1:300 for 45 min). Sections were washed three times with PBS after each antibody incubation. Dystrophin staining was observed on PBS-glycerol mounted slides under an ultraviolet lamp microscope using a fluorescein isothiocyanate filter.
Statistical Analysis
The significance of results was evaluated using Student's t-test between groups (Figs. 3 and 5) and ANOVA followed by Bonferroni correction of the t-test within groups (Fig. 4) on GraphPad software.

Improved differentiation of normal human myoblasts transduced with a follistatin lentivirus treated or not with recombinant myostatin. (a) Immunocytochemical detection of myosin heavy chain (in red) on human myoblasts, incubated for 2 days in differentiation medium, expressing eGFP or hFst and treated or not with recombinant myostatin (500 ng/ml), and corresponding nuclei stained with DAPI (100×). (b) Percentage of nuclei present in MyHC-positive cells over the total number of nuclei in normal human myoblasts transduced with eGFP or hFst lentivirus, and exposed or not to 150 ng/ml of recombinant myostatin (n = 3). ϕp < 0.01; *,τp < 0.05; φp < 0.1.
Results
Lentivirus Vector-Induced Follistatin Expression in 293T and Human Myoblasts
The ability of lentivirus vector to transduce human adult myoblasts was first assessed. The immunocytochemistry assay (Figs. 1b, 2a) showed that pCMV-eGFP transfected or transduced human adult myoblasts were follistatin negative, whereas pCMV-hFst transfected human myoblasts were all follistatin positive. The protein was thus well expressed in transduced cells. To confirm the results of the immunocytochemistry assay, a Western blot against follistatin was performed on a protein extract of each cell group (pCMV-eGFP or pCMV-hFst transfected 293T cells and pCMV-eGFP or pCMV-hFst transduced human myoblasts). The Western blots (Figs. 1c, 2b) clearly confirmed the overexpression of the follistatin protein by showing a more intense band in pCMV-hFst transfected 293T cells and transduced myoblasts in comparison with control cells.

Overexpression and secretion of follistatin in human myoblasts transduced with human follistatin lentivirus. (a, top) Normal human myoblasts (not fixed) expressing the eGFP after the lentivirus transduction (100×). (a, middle and bottom) Immunocytochemical detection of human follistatin in human myoblasts transduced with eGFP or with hFst lentivirus by and corresponding nuclei stained with DAPI (100×). (b) Western blot detection of human follistatin in proteins extracted from transduced human myoblasts. (c) Dot blot detection of human follistatin in conditioned media of transduced human myoblasts.
Overexpressed Follistatin Promoted Human Myoblast Proliferation In Vitro
For this experiment, myoblasts were first synchronized by serum deprivation for 48 h and complete medium was then added to the cells. Some of the cells died during the first 48 h due to the previous serum deprivation. The cells then proliferated. Figure 3 shows that the proliferation of pCMV-hFst transduced myoblasts was increased when compared with control myoblasts expressing eGFP. This could be explained by the fact that overexpressed follistatin antagonized the inhibitory effect of myostatin present in the proliferation medium.

Increased proliferation of human normal myoblasts overexpressing follistatin following lentiviral transduction. The kinetics of proliferation of control human myoblasts (transduced with eGFP lentivirus) in comparison with human myoblasts overexpressing follistatin protein based on a Cyquant cell proliferation assay. The result is reported as fluorescence percentage in comparison with the starting value corresponding to the fluorescence at T0 (100%) (n = 3). *p < 0.05.
Overexpressed Follistatin Promoted Human Myoblast Differentiation In Vitro
Recombinant myostatin blocked myoblast fusion and reduced the number of myotubes formed by eGFP and by hFst transduced myoblasts (Fig. 4). However, myoblasts expressing hFst formed more myosin heavy chain-positive myotubes than control eGFP-expressing cells. This is more evident in the absence of recombinant myostatin; indeed, when treated with the indicated dose of recombinant myostatin, hFst myoblasts failed to form significantly more myotubes than eGFP control cells. This could be explained by the fact that the amount of overproduced follistatin by hFst transduced cells is not sufficient to completely abolish the inhibitory effect of recombinant myostatin on differentiation process (Fig. 4).
Overexpressed Follistatin Enhanced the Success of Human Myoblast Transplantation in SCID Mice
Human myoblasts transduced either with eGFP or hFst lentivirus were transplanted in immunodeficient SCID mouse muscles to measure their potential to form muscle fibers expressing human dystrophin. An immunohistochemistry assay performed on sections of muscle transplanted either with GFP- or Fst-expressing human myoblasts (Fig. 5a) was used to count human dystrophin-positive fibers in each condition. Figure 5b shows that the success of the graft was clearly improved in muscles transplanted with myoblasts overexpressing the follistatin. Indeed, only 51 ± 14 hybrid fibers were counted in muscles transplanted with eGFP transduced myoblasts, while 106 ± 6 hybrid fibers were counted in muscle transplanted with hFst transduced myoblasts.
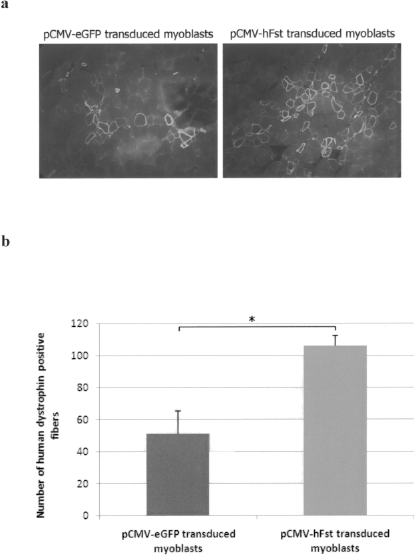
Improved transplantation success in SCID mice of follistatin-expressing myoblasts. (a) Representative sections of SCID muscles transplanted either with pCMV-eGFP transduced myoblasts, or pCMV-hFst transduced myoblasts and immunostained in red with an anti-human dystrophin antibody (100×). (b) Number of human dystrophin-positive fibers in SCID mouse muscles transplanted either with pCMV-eGFP or pCMV-hFst transduced myoblasts (n = 6). *p < 0.05.
Discussion
Normal myoblast transplantation in dystrophic muscle constitutes a potential therapy for Duchenne muscular dystrophy. However, the success of this potential therapeutic approach is limited by three major problems: 1) the poor survival of transplanted myoblasts, 2) the immune response against transplanted myoblasts, and 3) the fusion of the transplanted myoblasts only with the damaged muscle fibers located near the injection trajectories (28). In addition to those problems, the success of this therapy could be reduced by the short period of muscle regeneration, which follows muscle injury.
Myostatin is the most powerful inhibitor of muscle growth and regeneration identified to date (10, 17, 18). In fact, myostatin is known to regulate negatively both proliferation and differentiation of myoblasts, by the overexpression of the p21 cyclin-dependent kinase inhibitor (30), and also by downregulating the expression of muscle differentiation-related genes such as MyoD and myogenin (12). Myostatin also regulates muscle regeneration by blocking satellite cell activation and self-renewal. Myostatin action is antagonized by several proteins, including follistatin, which blocks its interaction with its specific receptor, ActRIIB (activin type II B receptor) (1). Moreover, follistatin regulates the expression of the myostatin gene in C2C12 myoblasts (11). In this study, we used a lentivirus to overexpress the short form of human follistatin in normal human myoblasts. An eGFP lentivirus was used as a control for all the experiments. The purpose of this work was to evaluate the effects of myostatin signal blockade by follistatin on proliferation, differentiation, and transplantation potential of myoblasts.
First, we demonstrated that adult human myoblasts were efficiently transduced with the follistatin-coding lentivirus and that these cells secreted this protein. Second, our results showed that the action of myostatin on myoblast proliferation and differentiation was blocked when follistatin was overproduced. This effect was probably mediated by the inhibition of the induction of p21 expression (9, 30) and the overexpression of MyoD (12, 23) in the absence of myostatin signal. This allowed myoblasts to progress in the cell cycle from the phase G1 and to differentiate in multinuclear myotubes. Finally, we assessed the potential of transduced myoblasts (eGFP or hFst) to form hybrid dystrophin-positive fibers when transplanted in immunodeficient SCID mouse muscles. The graft success was about twofold higher with hFst-expressing myoblasts than with control eGFP myoblasts. The improvement of myoblast graft success can be explained by the fact that both endogenous host myostatin and the myostatin produced by transplanted myoblasts themselves were inhibited by the overproduced follistatin protein. Thus, hFst myoblasts have better proliferation and differentiation rates than eGFP both in vitro and in vivo. However, the success of the myoblast transplantation, even with the myoblasts overexpressing the follistatin protein, was limited at around 100 fibers after the transplantation of 500,000 myoblasts. This could be explained by other limitations facing myoblast transplantation, such as the poor myoblast dissemination and survival (29).
In summary, myostatin fixation to the ActRIIB blocks myoblast proliferation and differentiation via three different intracellular pathways identified to date: Smads family of transcription factors (34), PI3K/Akt/GSK3β pathway (33), and finally the MAPK-dependent intracellular pathways (7, 22, 32).
We have shown here that combination of myoblast transplantation with induction of release of follistatin permitted to enhanced myogenic cell fusion and improved the graft success. By blocking the myostatin signal, follistatin could act by increasing and prolonging the regeneration period, which follows myoblast transplantation, and thus permits the fusion of the transplanted myoblasts with more host muscle fibers. It could also be responsible for the improvement of the dystrophic phenotype, as was shown recently by Minetti et al. (19). In addition, induced follistatin could positively act on myoblast graft success by reducing the fibrosis in transplanted muscle (14). The overexpression of follistatin is thus a procedure that may permit improving the success of myoblast transplantation as a treatment for Duchenne muscular dystrophy patients.
Footnotes
Acknowledgments
We thank Dr. Lee for giving us the follistatin plasmid. This work has been supported by the Association Française contre les Myopathies, the Canadian Institute for Health Research, and the Muscular Dystrophy Canada.