
Abstract
BACKGROUND:
Ciliary dysfunction underlies the pathogenesis of both heterotaxy syndrome and primary ciliary dyskinesia (PCD), often with overlapping genetic variants.
OBJECTIVE:
This case series aims to describe genetic testing and postoperative outcomes for infants with heterotaxy-associated congenital heart disease (H-CHD) with pathogenic variants in genes associated with ciliary structure or function.
METHODS:
Infants who underwent surgery for H-CHD between 2017 and 2022 were included in this single-center review. The results of genetic testing, microarray or sequencing-based tests, were reviewed. Baseline clinical data and postoperative outcomes are summarized for individuals with variants in ciliary genes.
RESULTS:
Of 32 infants who underwent surgery, 12 had sequencing-based testing. A genetic variant associated with ciliopathy was reported in 10 of 12 infants (83%), 3 (25%) were diagnostic of PCD and 2 (17%) were considered possibly diagnostic. Infants with variants in ciliary genes had high prevalence of postoperative respiratory complications, however a relationship between genetic test results and respiratory complications could not be proven. All infants with a genetic diagnosis of PCD showed clinical symptoms of PCD on follow-up.
CONCLUSIONS:
Sequencing-based testing has high detection rate for PCD in infants with H-CHD and may be valuable given their increased risk of respiratory complications after surgery.
Introduction
Heterotaxy syndrome describes a wide spectrum of congenital birth defects characterized by an abnormal lateralization of the visceral organs along the right-left axis. It is estimated to affect 0.01% of all live births and up to 3% of newborns with congenital heart disease (CHD) [1]. Infants with heterotaxy-associated congenital heart disease (H-CHD) tend to have complex anatomy that may include intracardiac defects and abnormal venous and arterial connections, resulting from disruption to the cardiac looping process in early embryogenesis. These infants frequently require high-risk surgical interventions, many of which follow a single ventricle palliation pathway (50%) or urgent postnatal repair of total anomalous pulmonary venous return (TAPVR, 38%) [2]. Studies showed that the anatomy, as well as the association with extra-cardiac anomalies, confer high morbidity and mortality for this specific group of patients, with no significant improvement in outcomes over the past three decades [3–5].
Primary ciliary dyskinesia (PCD) is a rare genetic disease characterized by impaired mucociliary clearance, chronic sinopulmonary infections, and progressive obstructive airway disease [6, 7]. In pediatrics, cardinal features include neonatal respiratory distress, chronic wet cough, chronic nasal congestion and associated airway defects [8]. Kartagener syndrome, the triad of situs inversus, chronic sinusitis and bronchiectasis, was associated with dysfunction of respiratory cilia in the 1970 s [9]. Embryologic cilia were implicated in the pathogenesis of situs inversus as their role in right-left lateralization in early embryogenesis was defined [10]. Additional work suggested that the spectrum of disorders known as heterotaxy is in fact a ‘ciliopathy’ in a similar manner, resulting from the disruption of embryonic ciliary structures [11, 12]. It was therefore suspected that variants in some genes that are shared across different ciliary structures could underlie both PCD and heterotaxy syndrome [13, 14]. The clinical significance of genetic variants in both disorders in predicting lung disease in infants with heterotaxy has not been well described. One challenge is that respiratory disease is multi-factorial and common in all infants with CHD who undergo cardiac surgery. Thus, it remains unknown to what degree respiratory ciliary dysfunction contributes to the higher morbidity in those infants with heterotaxy syndrome [15, 16].
As genetic testing has become more widely available for infants with complex CHD, it is our experience that infants with heterotaxy syndrome are often found to have variants in genes associated with PCD. In this study, we reviewed the results of genetic testing in those infants with H-CHD. We then evaluate reported variants in ciliary genes for a potential association with PCD and describe the respective cardiac anatomy and perioperative respiratory outcomes for each individual.
Materials and Methods
A single-center, retrospective case review was performed evaluating all infants diagnosed with heterotaxy syndrome and CHD at Seattle Children’s Hospital between January 2017 and June 2022. The study was approved by the Institutional Review Board at Seattle Children’s Hospital. The diagnosis of heterotaxy was obtained from clinical documentation or imaging reports. For all infants, both pre- and postnatal genetic testing results were reviewed. Testing included chromosomal microarray, panel-based testing, exome sequencing and genome sequencing. Test type was driven by recommendation of the clinician in discussion with each family, thus no unified guiding algorithm was used. Results were considered to be of interest if testing identified one or more variants in a gene encoding a protein known to be involved in ciliary structure or function. The pathogenicity of identified variants (variant of uncertain significance (VUS), likely-pathogenic, or pathogenic) was defined based on consensus guidelines [17]. As most causes of PCD are inherited in an autosomal recessive pattern, genetic testing was deemed diagnostic if both alleles of a ciliary gene harbored pathogenic or likely-pathogenic variants. The exception, in one case in this series, was a single pathogenic allele that was considered diagnostic based on previous evidence of autosomal dominant inheritance of PCD [18].
For infants with genetic test results of interest, review of the electronic medical record was performed for cardiac and respiratory diagnostic imaging, operative data, and perioperative outcomes. Cardiac and abdominal situs were determined by the reviewer based on the available imaging studies. Available computed tomography (CT) studies were reviewed to assess lung parenchyma and airway anatomy. For further PCD diagnostics, the results of nasal ciliary biopsies were reviewed if performed. No individual in the cohort underwent nasal nitric oxide (nNO) testing, as that is usually performed in older children [8]. Postoperative outcomes were assessed for the first surgery on cardiopulmonary bypass for each infant. At our institution the bidirectional cavopulmonary anastomosis surgeries (Glenn operations) are all performed while on cardiopulmonary bypass, and therefore included. Long-term follow up in Pulmonary Medicine Clinic was reviewed as of January 2023. All individuals were reviewed for the clinical diagnosis of pulmonary vein stenosis and pulmonary arterial hypertension. The hemodynamic data from pre- or postoperative cardiac catheterizations were collected to include relevant information on oxygenation and pulmonary blood flow, including mean pulmonary artery pressure, transpulmonary gradient, and indexed pulmonary vascular resistance, as a secondary marker of lung disease.
Results
Genetic Testing
Thirty-two infants with heterotaxy and CHD were identified over the study period (Fig. 1). Of these, 13 did not have any genetic testing performed and 7 had only microarray performed, all of which were negative. Twelve infants had sequencing-based genetic testing performed: panel-based testing (n = 5), exome sequencing (n = 4) or genome sequencing (n = 3). Of these, 10 infants had variants identified in cilia-related genes, 9 of whom had at least one pathogenic or likely pathogenic variant associated with PCD (Table 1). Variants in the dynein axonemal heavy chain family genes were the most prevalent and found in 7 of these 10 infants. Most infants who had sequencing-based genetic testing also had a microarray performed, either prenatally via amniocentesis (n = 4) or postnatally (n = 6). Four of these arrays had abnormal findings: with one providing a possible explanatory cause for CHD in an infant with no variants identified in ciliary genes by sequence-based testing and 3 infants with findings that were considered to likely be non-explanatory for CHD.
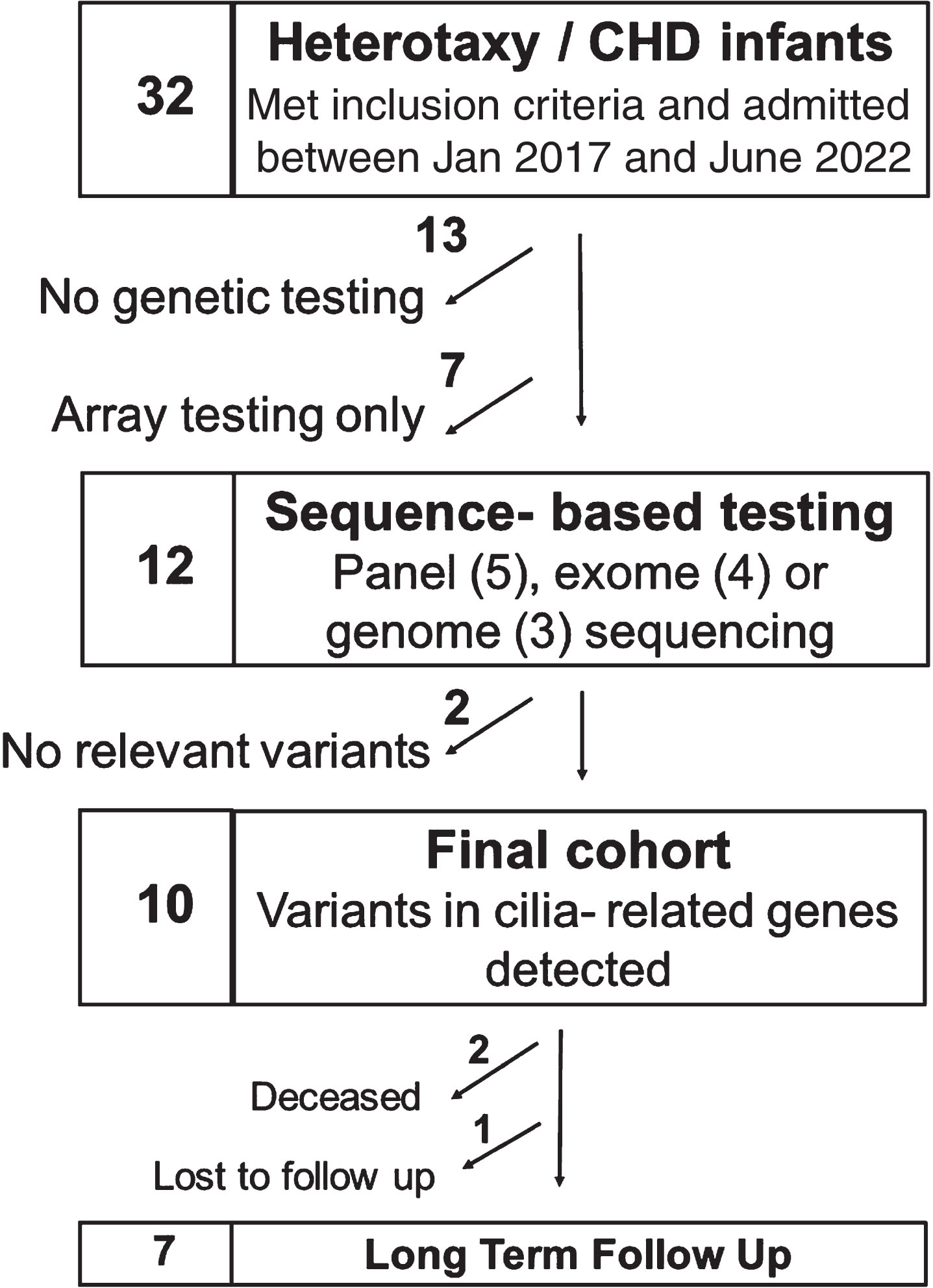
Consort diagram of patients’ genetic testing.
Phenotypes, Demographics, and Genetics for CCHD
Data is shown for patients with sequencing-based genetic and reported variants in genes related to PCD are shown. Allele frequency from gNomad v3.1.2. Abbreviations: TAPVR: total anomalous pulmonary venous return, SVC: superior vena cave, AVSD: atrioventricular septal defect, DORV: double-outlet right ventricle, MGA: malposed great arteries, HAA: hypoplastic aortic arch, ASD: atrial septal defect, AVV: atrioventricular valve, PA: pulmonary artery, IVC: inferior vena cava, VSD: ventricular septal defect, AVVR: AVV regurgitation, PV: pulmonary vein, GA: gestational age, M: male, F: female, PCD: primary ciliary dyskinesia, CHD: congenital heart disease.
A genetic diagnosis of PCD was made in 3 of 12 infants (individuals 1, 4 and 8). Two other infants were found to have either one pathogenic and one VUS in a ciliopathy gene (individual 7) or two pathogenic variants in a single gene, but we were unable to determine whether the two variants were on the same or different chromosomes (individual 10). For both infants, the available data is insufficient to make a genetic diagnosis, but the genotypes are possibly consistent with PCD. The clinical significance of genetic test results for the remaining 5 infants (2, 3, 5, 6, and 9) with variants in ciliopathy genes that are non-diagnostic of PCD remains unclear.
No correlation could be drawn between genetic diagnosis of PCD and lateralization arrangement (right or left isomerism), cardiac position, or cardiac morphology (Table 1). For specific complicating CHD factors: 7 infants out of 10 had ventricular hypoplasia and underwent single ventricle palliation, 6 had pulmonary valve stenosis or atresia and 2 had TAPVR. All 3 infants with a genetic diagnosis of PCD had single ventricle lesions with unbalanced atrioventricular septal defect. One infant included in this cohort had a VUS in three different genes (individual 5) and had both a perimembranous ventricular septal defect and secundum atrial septal defect.
The surgical history, as well as perioperative and follow up data, are summarized in Table 2. No correlation was suggested between parenchymal lung disease, postoperative outcomes and the genetic diagnosis of PCD. Chest CT studies were obtained for 8 of 10 infants. All infants had notable abnormalities, with diffuse atelectasis in all studies and ground glass opacities in 2 out of 8 (40%). With the exception of individual 5, all infants had a history of multiple postoperative respiratory complications and required respiratory therapies to assist in weaning off respiratory support. Five infants, all with single ventricle lesions, experienced very long hospital stays postoperatively (over 6 months), partly for prolonged respiratory support, or were readmitted at least once during their first year of life for respiratory infections. Four infants had pulmonary vein disease (with history of TAPVR at birth or vein stenosis acquired through the first year of life), all with single ventricle lesions. These infants all had cardiac catheterization performed (4 of 7 patients who had catheterizations performed) and had positive indices of pulmonary hypertension as expected. No additional correlation between pulmonary hemodynamics and a genetic diagnosis of PCD could be drawn.
Perioperative Outcomes
Perioperative Outcomes
Cardiorespiratory outcome measures summarized for patients with ciliary dyskinesia related genetic variants. Abbreviations: PV: pulmonary veins, AVV: atrioventricular valve, PA: pulmonary artery, mBTTS: modified Blalock- Thomas- Taussig shunt, ASD: atrial septal defect, VSD: ventricular septal defect, AVSD: atrioventricular septal defect, PVD: pulmonary vein disease, PAp: pulmonary arterial pressure, TPG: transpulmonary gradient, PVRi: indexed pulmonary vascular resistance, Wu: Woods units (for PVRi), CT: computed tomography, HFNC: high-flow nasal canula, CPAP: continuous positive airway pressure, iNO: inhaled nitric oxide, ECLS: extra-corporeal life support, PCD: primary ciliary dyskinesia, AW: airway.
Follow-up data after hospital discharge was available for 7 of 10 individuals, as 2 individuals in this cohort are deceased and one was lost to follow-up. The median age for follow-up was 21 months (range: 7–68). Four individuals were diagnosed clinically with PCD by a pediatric pulmonologist (BK). Of the 5 individuals with certain or possible genetic diagnosis of PCD, one is deceased and one was the youngest in our cohort and has yet to be seen by a pediatric pulmonologist after hospital discharge. The remaining 3 infants (individuals 1, 4 and 7) were clinically diagnosed with PCD. Those with a clinical diagnosis of PCD all benefited from airway clearance therapies for symptoms of congestion, heavy burden of airway secretions and frequent respiratory infections. Only 3 infants in this cohort underwent nasal ciliary biopsy, all of which demonstrated normal ciliary ultrastructure. These 3 infants did not have genetic diagnosis of PCD (individual 7 had a possible diagnosis by genetic testing). None of the children have yet to undergo nNO testing for PCD.
This case series outlines our single-center experience with the evaluation and perioperative care of infants with H-CHD, focusing on the subset of patients with genetic variants associated with ciliary structure or function. Our results suggest that sequencing-based testing for PCD is high-yield in infants with H-CHD for PCD. To our knowledge, this is the first report of genetic test results for infants with H-CHD [2]. While this case series is not designed to determine how genetic test results influence postoperative outcomes, the overall complexity and high morbidity of this cohort aligns with prior reports and emphasizes the need for further understanding of ciliopathies.
Sequencing-based genetic tests (panel-based, exome sequencing, or genome sequencing) have become more widely available and are now used more often in infants with complex CHD. This facilitates a genetic diagnosis of PCD in infants with heterotaxy syndrome. Sequencing-based testing revealed a high prevalence of positive results in this patient population as 3 out of 12 infants had genetic aberrations that meet diagnostic criteria for PCD, 2 infants had variants consistent with PCD but needed additional testing to fully resolve, and 5 additional infants had variants identified in genes associated with ciliary structure or function. This diagnostic yield (25%) was similar to previously reported cohorts of infants with heterotaxy syndrome, many of which used a single testing modality, or cohorts tested due to clinical suspicion of PCD [19–23]. Our institution does not have a consensus approach to genetic testing of infants with H-CHD and we observed a trend of using sequence-based testing more frequently over time.
Microarrays were sent for several individuals in our study, including 10 of the 12 that had sequencing-based testing, and were non-diagnostic in all but one case (Table 1). This suggests that in the absence of other compelling features, a sequencing-based test should be performed as first line in individuals with heterotaxy syndrome. Panel-based sequencing (compared to exome or genome sequencing), if not tested with parental samples, may make it difficult to properly phase (assign to maternal or paternal haplotypes) variants that were identified by sequencing leading to challenges with interpretation. For example, in individual 10, two pathogenic variants in DNAH11 were identified by a panel-based test without parental samples. The testing lab could therefore not determine whether the variants are on the same or different chromosomes, making it difficult to interpret the impact of both variants and confirm a diagnosis. We hope that in the future, newer testing modalities such as long-read sequencing, will allow phasing of variants in similar cases [24].
The cardiac diagnosis, as well as surgical timing and approach, varied significantly among individuals in this cohort, as expected [25]. Infants with TAPVR, critical pulmonary valve stenosis or atresia, or single ventricle anatomy, all required earlier, urgent or recurrent interventions [26, 27]. In this case series alone, we are thus unable to outline specific characteristics or prognostic variables as an outcome measure of genetic diagnosis of PCD. We also did not see a notable increase in pulmonary arterial pressures unless associated with a large left to right shunt or pulmonary vein disease. Our outcomes of death or orthotropic heart transplantation in 3 of 10 patients, in association with single ventricle palliation and pulmonary vein disease, is also consistent with previous reports, and no correlation with genetic testing for PCD was noted [5].
The infants in our cohort demonstrate a high rate of respiratory complications and prolonged hospitalization postoperatively, regardless of specific genetic testing results. All infants who had chest CT available for review showed structural abnormalities, including 2 of the 3 infants with biventricular circulation. As expected, these changes are multifactorial and influenced by surgical complications (such as diaphragm paresis) or abnormal airway anatomy. The phenotype of neonatal respiratory distress in PCD typically includes prolonged oxygen therapy and an increased frequency of lobar collapse [28]. Among those infants with chest CT data, all had evidence of bilateral atelectasis suggestive of lower airway obstruction. Parenchymal changes, notably, ground glass opacities, were also reported. In the setting of complex CHD, ground glass opacities could have various etiologies, including pulmonary edema from left-to-right shunting or elevated end-diastolic pressure. Still, studies have shown that postnatal lung fluid resorption is mediated by airway epithelium sodium channels (ENaC), which are found on respiratory ciliary surface [29]. This is hypothesized as a possible mechanism for PCD neonatal distress and could possibly be a contributing factor to parenchymal lung disease in heterotaxy as well.
In our cohort, nasal biopsies were performed on only 3 infants and all of which were normal. While it is a recognized limitation, given that PCD is typically clinically diagnosed through functional testing of respiratory cilia, our review focused on genetic testing. Previous studies in CHD population demonstrated a high rate of abnormal ciliary function by nasal biopsy or nNO testing that associated with postoperative respiratory complications [30–32]. The 3 infants in our cohort who had nasal biopsies all did not have a genetic diagnosis of PCD, but two were treated as PCD on follow up. We feel that better understanding of genetic testing results is particularly important in evaluating for PCD as other testing modalities, such as nNO testing, may be impractical and insensitive in the preoperative evaluation of the young infant with H-CHD and have no role in prenatal counseling [8, 33].
Finally, long-term follow up suggests that infants with a genetic diagnosis of PCD have respiratory symptoms consistent with PCD and benefit from respiratory therapy for airway clearance. PCD was shown to be associated with prolonged length of stay and decreased lung function on follow up in neonates without CHD [34]. These clinical implications emphasize, again, the importance of early diagnosis through genetic testing and understanding of the genetic diagnosis of ciliary dysfunction for infants with H-CHD.
Limitations
This report has the inherent limitations of a single-center retrospective case review. It is also limited by the small number of subjects as it reports a rare clinical condition. Selection and test bias are both recognized in this report with no consensus approach to the choice of genetic testing per patient and variation during the study period. This study did not establish a matched control group given the small and heterogenous nature of the cohort. Data on clinical outcomes is therefore observational only and does not determine a causative effect for PCD diagnosis and respiratory complications. Also, as most individuals included in this study are still young, long-term follow-up on clinical outcomes and functional testing for PCD is lacking. We hope that collaborative multi-center work will be funded to study the clinical significance of genetic variants in PCD- related genes in H-CHD.
Footnotes
Acknowledgments
No external funding.
Author Contributions
JM: conception, analysis of data, preparation of manuscript
LNC: analysis of data, preparation of manuscript
DEM: analysis of data, revision for important intellectual content
MDF: revision for important intellectual content
BK: conception, analysis of data, revision for important intellectual content
ES: conception, analysis of data, preparation of manuscript, supervision
Conflict of interest
Dr. Miller holds stock options in MyOme, is engaged in a research agreement with Oxford Nanopore Technologies, and they have paid for him to travel to speak on their behalf. All other authors declare that they have no conflict of interest.