
Abstract
Joubert syndrome (JS; MIM PS213300) is a rare, typically autosomal recessive disorder characterized by cerebellar vermis hypoplasia and a distinctive malformation of the cerebellum and brainstem identified as the “molar tooth sign” on brain MRI. Other universal features include hypotonia with later ataxia and intellectual disability/developmental delay, with additional features consisting of oculomotor apraxia and abnormal respiratory pattern. Notably, other, more variable features include renal cystic disease, typically nephronophthisis, retinal dystrophy, and congenital hepatic fibrosis; skeletal changes such as polydactyly and findings consistent with short-rib skeletal dysplasias are also seen in many subjects. These pleiotropic features are typical of a number of disorders of the primary cilium, and make the identification of causal genes challenging given the significant overlap between JS and other ciliopathy conditions such as nephronophthisis and Meckel, Bardet-Biedl, and COACH syndromes. This review will describe the features of JS, characterize the 35 known genes associated with the condition, and describe some of the genetic conundrums of JS, such as the heterogeneity of founder effects, lack of genotype-phenotype correlations, and role of genetic modifiers. Finally, aspects of JS and related ciliopathies that may pave the way for development of therapeutic interventions, including gene therapy, will be described.
Keywords
Introduction
Joubert syndrome (JS; OMIM PS213300) is a predominantly autosomal recessive ciliopathy condition characterized by a distinctive cerebellar and brainstem defect on cranial MRI known as the “molar tooth sign” because of its resemblance to the cross-section of a tooth on axial imaging [64, 65]. Its three key diagnostic criteria have been established as (1) the “molar tooth sign” (MTS), (2) hypotonia in infancy with later ataxia, and (3) developmental delays/intellectual disability. There are several other more variable features, including breathing problems— often episodic tachypnea and/or apnea— especially in infancy, and abnormal eye movements typically characterized by oculomotor apraxia or the inability in initiating voluntary saccades [70]. In addition, a number of findings are sometimes found in those with JS such as retinal disease, cystic kidney disease including nephronophthisis, congenital hepatic fibrosis, and skeletal features such as polydactyly and small ribcage; many of these are also shared by other ciliopathy conditions [6, 118].
Since the first gene for JS, NPHP1, was identified in 2004 [72], over 30 genes have been implicated in its causation [70]. However, most of these genes cause less than 10% of cases each, and for several genes, the assignment is based on only one or a handful of cases. All of the genes identified thus far are localized to or play a role in the function of the sub-cellular structure, the primary cilium, particularly at the transition zone of the cilium where it joins the plasma membrane [36]. The primary cilium is the appendage on almost all cells that functions in the cell’s ability to establish a pattern of left-right symmetry during development, allows it to transduce signals from the environment, and carries out fundamental intracellular processes via signaling pathways such as Sonic Hedgehog and others [82].
This review will focus on the molecular genetics of JS, while also including genetics-based discussion of other relevant ciliary disorders with overlapping phenotypes, and will provide some examples of potential future treatments to capitalize based on what is known about gene function and ciliary biology.
Diagnostic criteria for Joubert syndrome
The diagnosis of JS has been established as requiring 3 key elements: the radiologic finding of the “molar tooth sign” (MTS; Fig. 1A, C) accompanied by cerebellar vermis hypoplasia, as well as hypotonia or low muscle tone and developmental delays or intellectual disability. The MTS is visualized on axial views from magnetic resonance imaging (MRI) studies through the junction of the midbrain and pons and consists of the mesencephalon (the “crown” of the tooth), typically with a deepened interpeduncular fossa, and prominent, thickened, and generally straight superior cerebellar peduncles (the “roots” of the tooth) [64, 65]. The MTS is generally associated with hypoplasia of the midline portion of the cerebellum (the cerebellar vermis), a required feature in JS. On sagittal MRI, an enlarged, misshapen and/or rostral shifting of the fastigium of the 4th ventricle is often visualized (Fig. 1B, D). Although a high-resolution MRI scan with 3 mm sections is recommended for visualizing this radiologic sign, the MTS can sometimes be viewed on good quality, high density computed tomography (CT) scans. In reality, a radiologic finding of the MTS is often considered “pathognomonic” for JS, and certainly this discovery in the prenatal or neonatal period would warrant a presumptive diagnosis of JS, as the other required features are likely to become apparent with time (see refs [27, 80] for prenatal radiologic features in JS). In addition, there is a spectrum of MTS findings, with milder versions seen in association with some causative genes [31, 72], and without examination of multiple MRI cuts for subtle findings of the MTS and vermis hypoplasia, even experienced neuroradiologists may miss this diagnostic hallmark.
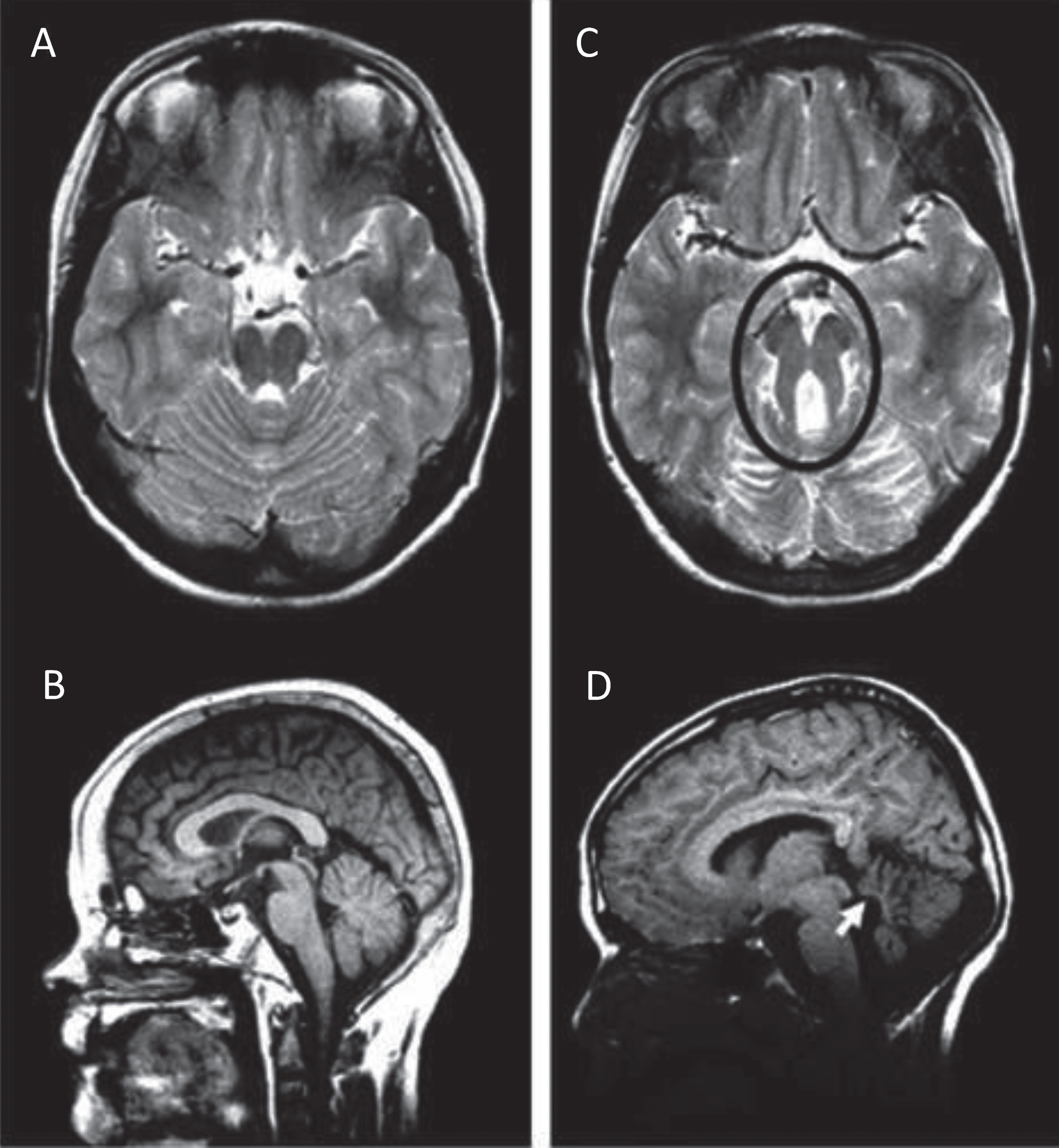
Molar Tooth Sign in Joubert syndrome. T2-weighted MRI images on axial view (A) and mid-sagittal view (B) through the cerebellum and brainstem of a normal individual showing intact cerebellar vermis. T2-weighted MRI image on axial view (C) and mid-sagittal view (D) through the cerebellum and brainstem of a child with JS. The Molar Tooth Sign is circled in black, with rostral shifting or elevation of the roof (fastigium) of the fourth ventricle and vermis hypoplasia (white arrow).
The hypotonia of infancy often gives way to later ataxia. Developmental delays in motor and cognitive development are universal [76, 105], although rare individuals with JS and normal IQ have been described [76]. Of note, a correlation between degree of cerebellar vermis hypoplasia and cognitive impairment has been seen [77]. In fact, a systematic evaluation of the neuropsychological profiles in 76 participants with JS showed an average Full Scale Intelligence Quotient (FSIQ) = 64±15 and relative strengths in receptive language and verbal comprehension, but challenges with processing speed most likely related to cerebellar deficits [105]. Behavioral surveys suggested more emotional and behavioral problems than in the general population, which may worsen with age, but not as severe as other populations with intellectual disability [105].
Although the 3 cardinal features are necessary for a diagnosis, additional features are often described in infants and children with JS. An abnormal respiratory pattern, particularly prominent in infancy, consists of alternating tachypnea and/or apnea; some neonates have had worrisome bouts of apnea requiring pharmacological intervention [63, 103]. In fact, for those with JS under 5 years of age who were deceased, respiratory failure was the leading cause of death [24]. Eye movements characterized as oculomotor apraxia, or the absence of or defect in controlled, voluntary, and purposeful eye movements resulting in jerky eye movements and head thrusting, as well as nystagmus, strabismus, and ptosis of the eyelids, are often identified in those with JS (Fig. 2A). The spectrum of ocular findings in JS has been prospectively ascertained in one large cohort [16] and the published literature on ocular findings in JS has also been reviewed recently by two groups [16, 119], with general findings of delayed visual development and variable visual acuity [16, 119]. Retinopathy has been reported in nearly 1/3, and ocular coloboma in nearly 30%; however, these rarely occur in the same individual. Optic nerve atrophy was reported in only 10–15% of these cohorts [16, 119].
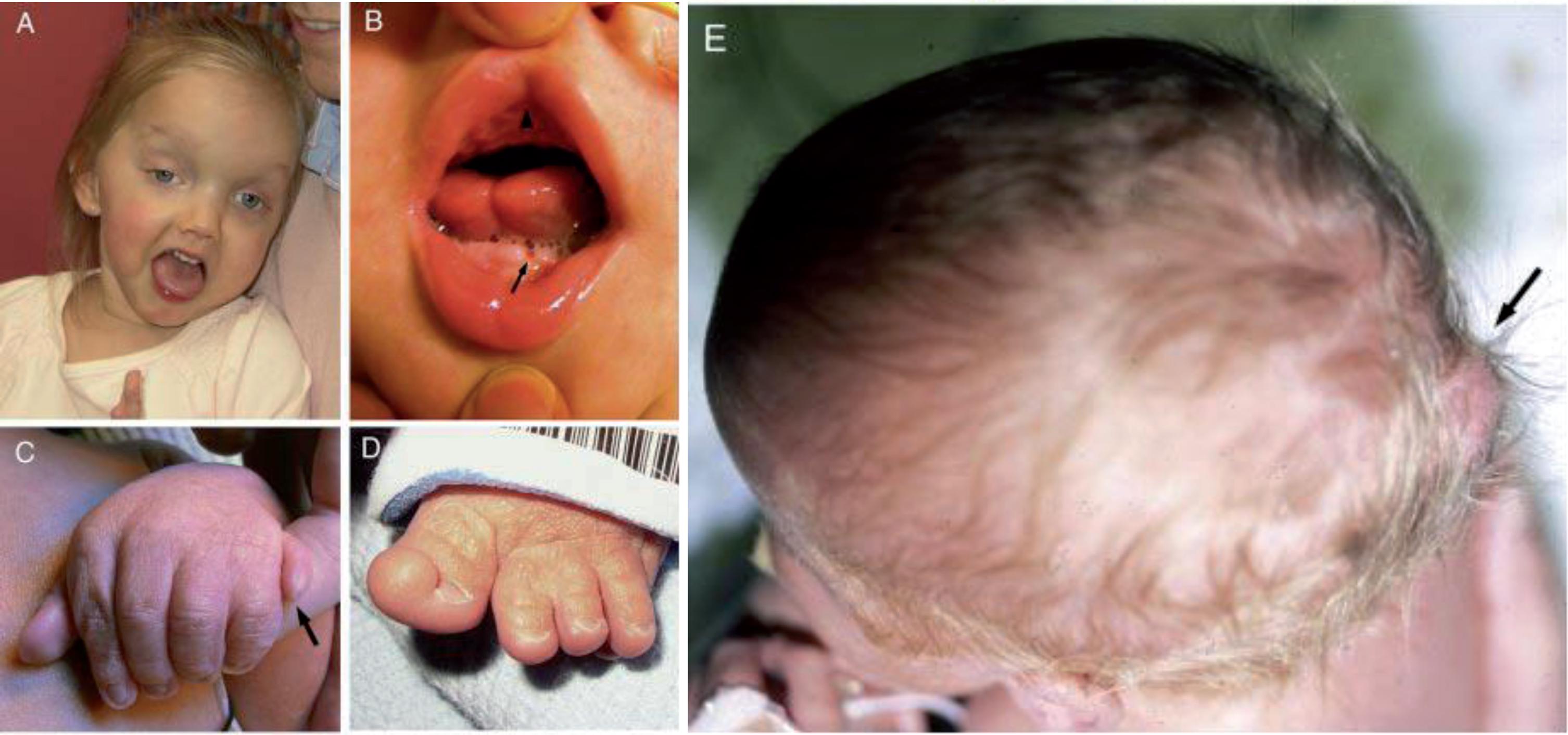
Clinical features in Joubert syndrome. A. Facial features in a girl with JS/COACH syndrome at 27 months of age showing broad forehead, arched eyebrows, strabismus, eyelid ptosis (on right eye), and open mouth configuration indicating reduced facial tone. B. Oral findings in a child with oral– facial– digital syndrome-like features of JS showing midline upper lip cleft (arrowhead), midline groove of tongue, and bumps of the lower alveolar ridge (arrow). C. Left hand of an infant with JS and postaxial polydactyly (arrow). D. Left foot of an infant with JS and preaxial polysyndactyly and fusion of the hallux. E. View from above of an infant with a small occipital encephalocele with protrusion of the occiput of the skull (arrow). (Facial photograph used with permission of the family.) (Previously published in [73] with permission).
Phenotypic variability is a key component of JS. There are pleiotropic and highly variable features described in other organ systems that can provide clues to sub-phenotyping, and in some cases, even hints at underlying genetic causes. In addition to the eye findings described above, significant morbidity can arise in those with JS and renal disease (nephronophthisis or cystic kidneys) or congenital hepatic fibrosis. Renal failure is the leading cause of death in those over the age of 5 years [24]. In addition, they can have oral or tongue hamartomas (Fig. 2B), skeletal changes (polydactyly (Fig. 2C, D), syndactyly, and/or narrow thoracic ribcage), and occipital encephalocele (Fig. 2E) [6, 118]. The polydactyly can be postaxial (Fig. 2C), preaxial (Fig. 2D), and occasionally central or mesaxial. Congenital heart defects, cleft palate, skin findings related to those of cranioectodermal dysplasia (sparse hair, small, widely spaced teeth or hypodontia, and/or abnormal nails), situs defects, and obesity are less common but occasionally described. Although ocular colobomas and occipital encephaloceles are developmental defects present at birth, many other features evolve over time and with age, so regular monitoring is necessary to ensure timely diagnosis and treatment if appropriate [70]. Many of these features are typical in other ciliopathy disorders such as Meckel syndrome (MKS) or Oral-Facial-Digital (OFD) syndrome, reinforcing the overlapping phenotypic spectrum of these conditions and confounding attempts at tidy genotype-phenotype correlations [114].
Given the broad clinical heterogeneity of Joubert syndrome, there are some useful sub-categories that can help in classifying the condition, and sometimes, in predicting which additional manifestations may develop in a given individual [14]. However, even within the same family, siblings with the same causative mutations may have variable phenotypes, illustrating that clinical heterogeneity is the norm in this condition.
Pure or classic JS
In this category, “pure” or “classic” JS has the 3 main diagnostic findings and often includes oculomotor apraxia and/or respiratory abnormalities, and may or may not be associated with polydactyly and other less common manifestations [70]. Many children born with JS are initially considered in this category, until they exhibit manifestations of retinal and/or renal or hepatic disease with age.
JS with retinal disease (JS-Ret)
This form of JS is associated with pigmentary retinopathy that resembles classic retinitis pigmentosa (RP). In some newborns, the most severe manifestations can resemble congenital blindness with attenuated electroretinogram in the spectrum of Leber Congenital Amaurosis (LCA) [16]. The retinal degeneration may develop with age but may not be as rapidly progressive as in some genetic forms of RP [103]. Individuals with retinal disease rarely have ocular colobomas [16]. Overall, retinal dystrophy has been reported in 24–32% of individuals with the condition [16, 28] and has been strongly linked to the AHI1, ARL3, and CEP290 genes [3, 119].
JS with renal disease (JS-Ren)
The kidney disease in JS is generally one of two forms: nephronophthisis (NPHP) or cystic kidneys, although both of these can be part of a spectrum of cystic renal diseases. NPHP manifests as chronic tubulointerstitial nephropathy, and may present in the first or second decade of life with urine-concentrating defects and increased thirst, often progressing to end-stage renal disease (ESRD) within a decade. In juvenile NPHP, the most typical form in JS, ESRD occurs at a median of 13 years [45]. Pathological evaluation of the kidneys reveals corticomedullary cysts, atrophy and interstitial fibrosis; ultrasound evaluation may demonstrate small, scarred kidneys with increased echogenicity at the corticomedullary junction [33, 99]. Another form of kidney disease manifests as enlarged kidney with multiple cysts resembling autosomal recessive polycystic kidney disease, with attendant risk of hypertension [33, 41]. In several surveys, between 23–32% of those with JS have renal manifestations [26, 118].
JS with oculorenal disease (JS-OR)
The combination of kidney disease and retinopathy is often seen in the same person and is associated with a number of genes that cause JS. Historically, the combination of RP and nephronophthisis was known as Senior-Løken syndrome [14, 71] and has been reported in about a third of those with kidney disease [118].
JS with hepatic disease (JS-H)
JS with hepatic disease is rarely symptomatic at birth, although it is characterized by congenital hepatic fibrosis, a developmental disorder of the portobiliary system caused by dysfunctional cilia. The related ciliopathy, Meckel syndrome, also has congenital hepatic fibrosis with ductal plate malformation as a consistent finding [40]. Features include persistence of embryonic ductal plate structures, cystic dilation of intrahepatic biliary ducts, and progressive fibrosis of the portal tracts [104]. Symptoms are generally related to portal hypertension, and individuals can present with elevated liver enzymes, or with advanced disease, recurrent cholangitis and/or gastroesophageal variceal bleeding with thrombocytopenia. Liver disease is often associated with ocular coloboma, a developmental defect of the eye, and can also be associated with kidney disease as well.
JS with oral-facial-digital features (JS-OFD)
Oral features include midline cleft lip and/or palate, midline groove of the tongue, gum or tongue hamartomas, and oral frenulae (Fig. 2B). Other facial features include hypertelorism and micrognathia or recessed jaw. The polydactyly in JS is often postaxial (Fig. 2C) although preaxial polydactyly of the great toes is not uncommon (Fig. 2D); it can be found in any combination on hands and/or feet [70]. Overall, polydactyly has been described in 13–15% of individuals with JS [6, 118]. These features are typical in the genetically heterogeneous group of ciliopathies known as oral-facial-digital (OFD) syndromes. A distinctive form of midline polydactyly known as mesaxial polydactyly and associated with a Y-shaped metacarpal is described in a specific type of OFD known as OFD type VI [12, 37].
JS with acrocallosal features (JS-AC)
In addition to the described structural cerebellar malformations in the human brain in JS, some affected individuals also have abnormalities of the corpus callosum, which connects the two cerebral hemispheres. On careful inspection of MRI scans, one survey found that up to 80% of JS subjects had some sort of callosal dysgenesis [92]. Some families with JS-AC features have mutations in the KIF7 gene, which also causes acrocallosal syndrome with polydactyly and hydrocephalus [6, 79].
JS with Jeune asphyxiating thoracic dystrophy (JS-JATD)
Skeletal abnormalities described in short-rib polydactyly phenotypes include narrow thoracic rib cage, short ribs, shortened tubular bones, and a ‘trident’ appearance of the acetabular roof, with or without polydactyly. Similar skeletal features have been described in subjects with JS and the MTS, most typically, short stature, narrow rib cage, rhizomelic limb shortening, cone-shaped phalangeal epiphyses, brachydactyly, and polydactyly [42, 95].
Prevalence and epidemiology of Joubert syndrome
The prevalence of JS has been estimated to be between 1:80,000 and 1:100,000 [14, 73], but this may represent an underestimate as more causative genes and private mutations are identified, as well as a broader range of phenotypic findings. JS is a predominantly autosomal recessive disorder first described in relatively isolated groups with high rates of consanguinity, including in French-Canadian, Ashkenazi Jewish, and Arab populations [30, 100–102]. Interestingly, there are multiple founder mutations in several genes for each of these populations. The original French-Canadian family described by Joubert et al in 1969 [49] was known to have distant consanguinity related to a common founder 10 generations removed in France. However, there are at least 3 different genes known to cause JS in families from this region of Canada (CPLANE1, CC2D2A, NPHP1), and the breadth of mutations even within genes linked to an inbred population is highly variable. In fact, many affected individuals who are French-Canadian harbor heterozygous pathogenic variants [100], indicating that the concept of a single “founder” mutation in a particular consanguineous population does not apply to JS. This phenomenon also applies to families from the Middle East, where although there is a high rate of consanguinity, there is a diversity in the number of causative genes [6]. However, in many families of relatively close Arab descent, with first cousin or comparable relatedness, the pathogenic variants are likely to be homozygous, reflecting identity-by-descent. The heterogeneity of causative genes, and in some cases, mutations in an inbred population has also been reported in other ciliopathy disorders. In Newfoundland, for example, multiple different founder effects appear to be causative of Bardet-Biedl syndrome (BBS; [67]). A plausible molecular explanation has not been postulated.
Genes associated with Joubert syndrome
There are 35 genes implicated as causative for JS (Table 1). Although most are inherited in an autosomal recessive manner, as are the majority of ciliopathies, one, OFD1 (Oral Facial Syndrome 1) demonstrates X-linked recessive inheritance. A few genes have been suggested to have autosomal dominant inheritance, but this is highly speculative as exhaustive screening for other variants may not have been performed. Collectively, these genes account for between 62–94% of cases of JS [6, 118]. The high rate of 94% with identified genetic causes in one survey [118] may be related in part to the large proportion of subjects with hepatic involvement and COACH phenotypes, with better known and characterized genetic causes. Of note, almost all of the genes have also been implicated in other ciliopathy disorders, such as MKS, NPHP, OFD, BBS, COACH syndrome, and others (see column of “Allelic disorders” in Table 1). In general, it is difficult to identify clear-cut genotype-phenotype correlations for many of these genes, but a few distinguishing features are included in the table, as well as genes where founder effects have been identified. In some cases, a more severe or even lethal condition, such as MKS, has been associated with more severe pathogenic changes such as frameshift or nonsense mutations causing loss-of-function rather than milder missense variants often associated with a non-lethal phenotype, such as JS [14]— See Additional Challenges below.
Genetic causes of Joubert syndrome, listed in ascending order by chromosomal locus, starting with chromosome 1
Genetic causes of Joubert syndrome, listed in ascending order by chromosomal locus, starting with chromosome 1
General references for these tables include [38, 82] and OMIM. 1For those genes that account for >1% of JS cases in several large surveys ([6, 118]), the frequency is noted. 2Dominant loss-of-function mutations in the SUFU tumor suppressor gene are associated with medulloblastoma, Basal Cell Nevus Syndrome, and susceptibility to familial meningioma. 3Pathogenic variants in this gene have been identified in conjunction with the MTS, but this gene has not been formally assigned an OMIM designation in the JS phenotypic series PS213300. This status also applies to the following genes: C2CD3, HYLS1, KIAA0753, IFT80, and CELSR2. Abbreviations: ACC, agenesis of the corpus callosum; ACS, Acrocallosal syndrome; AD, autosomal dominant; AR, autosomal recessive; BB, Basal body; BBS, Bardet-Biedl syndrome; BCNS, Basal Cell Nevus Syndrome; COACH, Colobomas, cognitive impairment (“Oligophrenia”), Ataxia, Cerebellar vermis hypoplasia, and Hepatic fibrosis syndrome; CP, cleft palate; CRD, Cone-rod dystrophy; ENC, encephalocele; HTN, hypertension; JATD, Jeune asphyxiating thoracic dystrophy; JS, Joubert syndrome; LCA, Leber Congenital Amaurosis; MKS, Meckel syndrome; MORM, Mental retardation, Obesity, Retinal dystrophy, Micropenis syndrome; NPHP, nephronophthisis; OFD, oral-facial-digital syndrome; PD, polydactyly; PIT, pituitary abnormalities, including hypopituitarism, micropenis; PMG, polymicrogyria; RP, retinitis pigmentosa; SLS, Senior-Løken syndrome; TZ, transition zone; XLR, X-linked recessive.
Ciliopathy conditions are human disorders characterized by dysfunction of either the motile or immotile cilium, a subcellular organelle important for embryonic development, adult homeostasis, and in sensory and/or signaling processes [38]. JS is a ciliopathy disorder of the primary, non-motile cilium found on almost all vertebrate cells [82]. Most of the genes implicated in JS and related ciliopathies localize to either the primary cilium, the basal body and/or the mother centriole from which it arises, or the regulatory proteins and transcription factors that guide its development and function [82]. The primary cilium has been described as an antennae or appendage protruding from the plasma membrane that plays a role in sensing the environment and responding to localized signals [38]. It develops from the docking of a mother centriole via “distal appendages” or transition fibers that link the centriole to the plasma membrane; this is where the centriole extends the 9 axonemal microtubule doublets into the cilium (Fig. 3A, B). The structure at the base of the cilium and junction of plasma membrane and ciliary membrane is known as the transition zone [36]. This “transition zone,” which, along with the transition fibers and Y-fibers, comprise the “ciliary gate,” lies between the basal body and the ciliary axoneme and controls the entry and exit of proteins and lipids into and out of the cilium. Intraflagellar transport (IFT) of this cargo along the microtubules is driven by kinesin (anterograde) and dynein (retrograde) motors and associated IFT proteins and BBS proteins, with lipidated protein transport also playing a role [82].
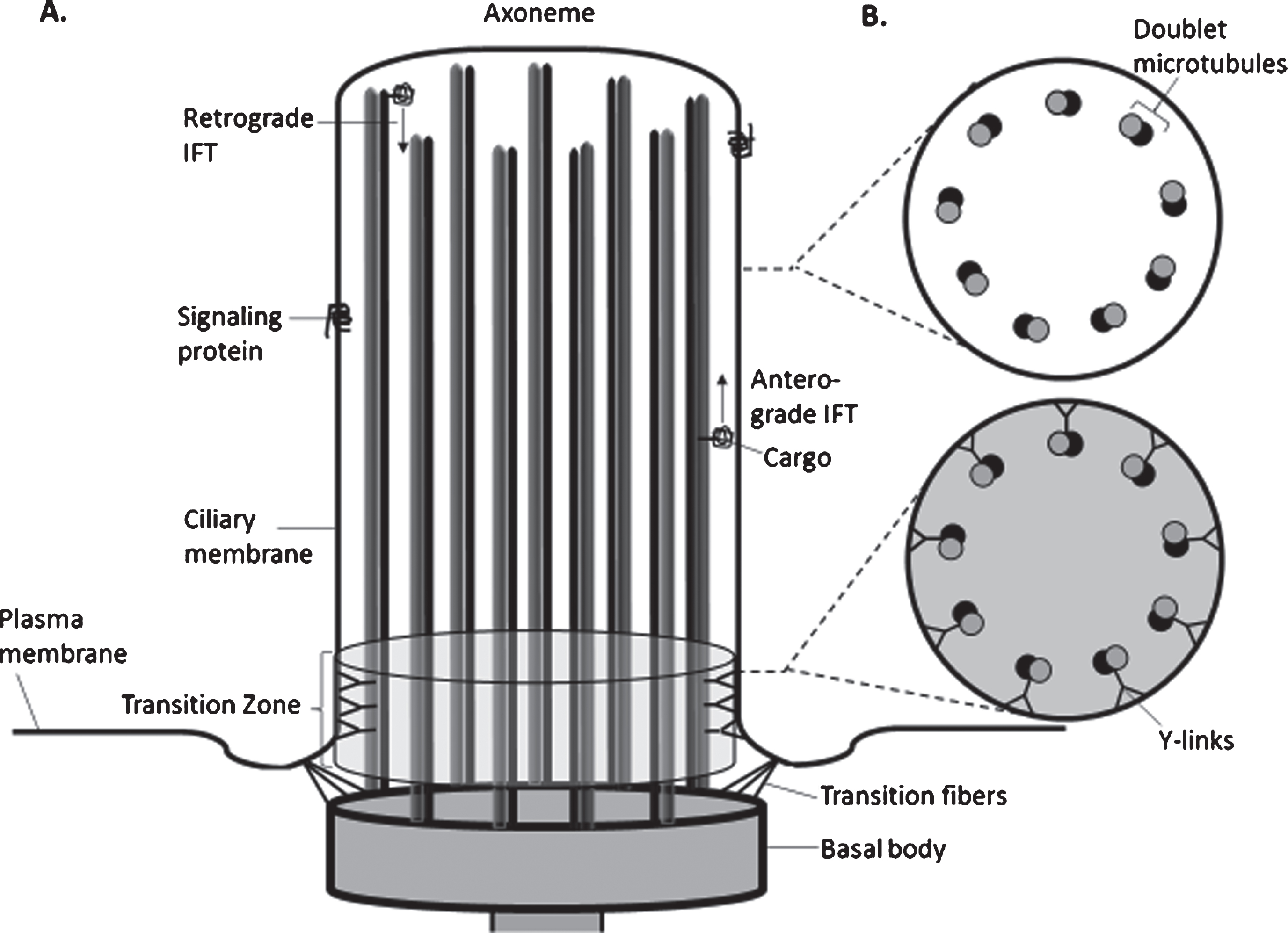
Diagram of the structure of the Primary Cilium. A. Schematic of the components of a non-motile (primary cilium) showing the axoneme (ciliary stalk), comprised of 9 pairs of microtubules. The cilium extends from a basal body, which derives from the mother centriole. Transition fibers tether the basal body to the base of the ciliary membrane. The transition zone (TZ) is adjacent to the basal body and serves as a gate for passage of proteins and lipids into and out of the cilium. Anterograde transport is accomplished by kinesin motors along with associated proteins and BBsomes propelling cargo along the microtubules; retrograde transport utilizes dynein motors rather than kinesins. Signaling proteins on the ciliary membrane represent the components of the Sonic Hedgehog (SHH) or other receptor complexes. B. Cross-section of the cilium demonstrates the typical 9+0 pattern of doublet microtubes in the axoneme (upper diagram) and the addition of Y-links that connect the doublet microtubes to the ciliary membrane and help form the barrier within the transition zone (bottom diagram). (Derived from [38, 82]).
The transition zone appears to be a hotspot for human disease genes associated with ciliopathies. In fact, there are either 2 or 3 main protein complexes, mapped by interactome studies, that form the transition zone [19, 35]: the MKS complex, which includes the Tectonic proteins, B9 domain proteins, coiled-coil proteins such as CC2D2A and CEP290, AHI1, and 5–7 transmembrane or TMEM proteins; and the NPHP complex, including the NPHP proteins, RPGRIP1 L, and others [36]. Linking these 2 domains are CEP290 and NPHP5, sometimes separated out as a distinctive protein domain [38]. These subdivisions are significant, as almost all of the proteins in the
JS is one of several disorders of the primary cilium that have multi-systemic effects, often including involvement of the brain, eyes, kidney, liver, and skeleton. These ciliopathies also overlap in their manifestations, and in their causative genes [66]. The allelic ciliopathy disorders associated with each of the 35 JS-associated genes based on overlapping phenotypes are shown in Fig. 4. The following sub-sections will describe the most common allelic disorders to JS identified thus far:
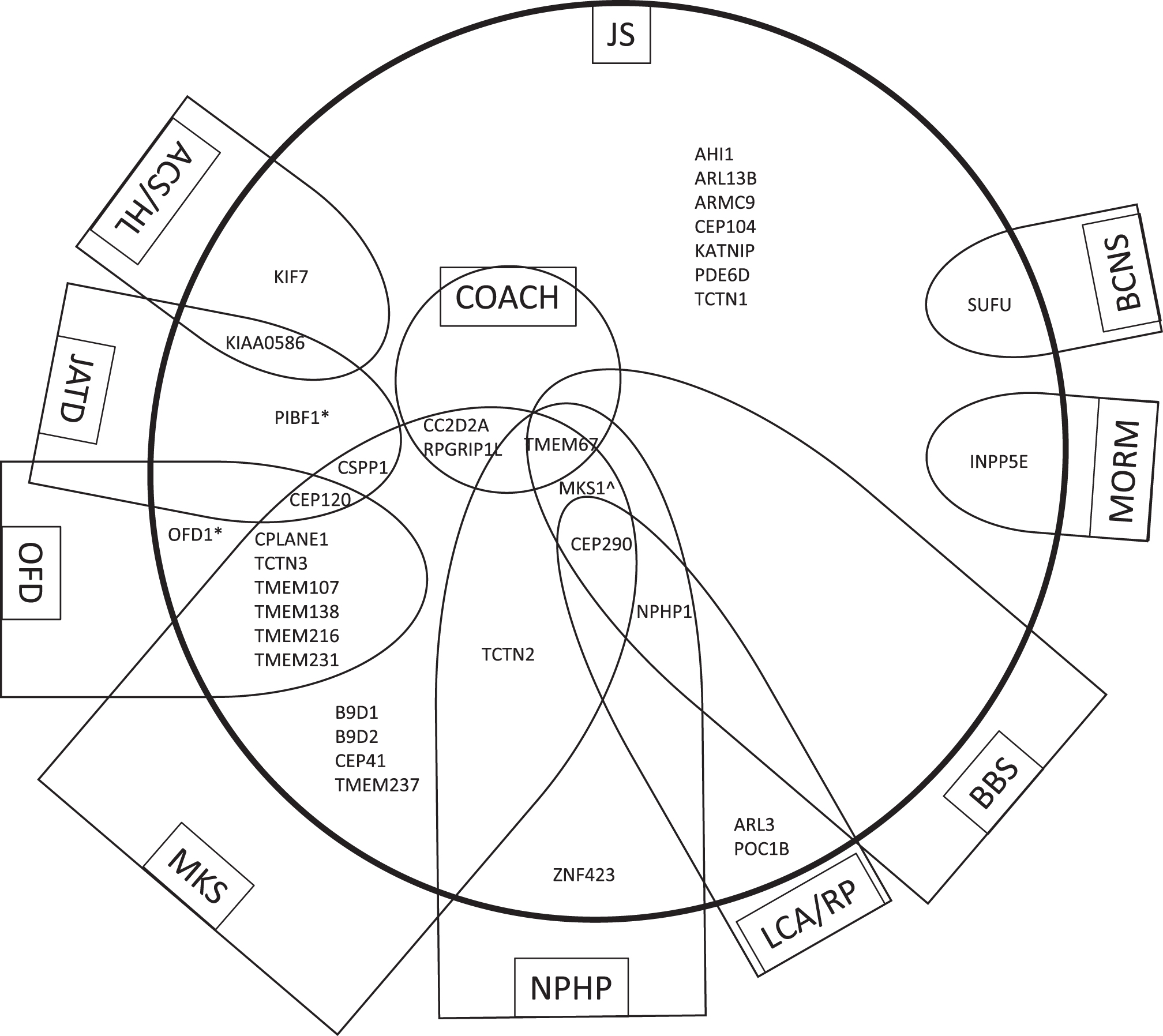
Genes that cause JS have overlapping phenotypes with other ciliopathies. The 35 genes that cause Joubert syndrome (JS) and are listed in Table 1 are distributed within the large circle in relationship to allelic ciliopathy disorders indicated within each semi-oval (or circle in the case of COACH). The name of each ciliopathy disorder is indicated within the box. Other genes that cause the ciliopathy condition but are not associated with JS are omitted in order to simplify the diagram. These relationships are not fixed, as the phenotypic spectrum associated with ciliopathy genes is constantly evolving. *Can also be associated with RP alone ∧The MKS1 gene has not been described in NPHP alone but this could not be indicated due to constraints of the diagram. Abbreviations: ACS, Acrocallosal syndrome; BBS, Bardet-Biedl syndrome; BCNS, Basal Cell Nevus Syndrome; COACH, Colobomas, cognitive impairment (“Oligophrenia”), Ataxia, Cerebellar vermis hypoplasia, and Hepatic fibrosis syndrome; HL, Hydrolethalus syndrome; JATD, Jeune asphyxiating thoracic dystrophy and related skeletal dysplasias; JS, Joubert syndrome; LCA, Leber Congenital Amaurosis; MKS, Meckel syndrome; MORM, Mental retardation, Obesity, Retinal dystrophy, Micropenis syndrome; NPHP, nephronophthisis; OFD, oral-facial-digital syndrome; RP, retinitis pigmentosa.
Acrocallosal syndrome (ACLS; OMIM 200990) is a recessive condition with macrocephaly, agenesis of the corpus callosum, other brain anomalies, intellectual disability, and polydactyly. The previously reported association in some families with KIF7 mutations and either an ACLS, JS or
Bardet-Biedl syndrome (BBS, OMIM PS209900)
Bardet-Biedl syndrome (BBS, OMIM PS209900) is generally autosomal recessive in inheritance, with some examples of digenic inheritance, and is characterized by cone-rod retinal dystrophy, obesity, postaxial polydactyly, intellectual disability, variable kidney disease, and hypogonadism/genital anomalies. Progressive renal and retinal disease can be a source of morbidity and mortality; at least 19 genes have been implicated thus far [34].
COACH syndrome (COACH, OMIM 216360)
COACH syndrome (COACH, OMIM 216360) the combination of
Jeune asphyxiating thoracic dystrophy (JATD)
Jeune asphyxiating thoracic dystrophy (JATD) is one of a group of autosomal recessive skeletal dysplasias known as short-rib thoracic dystrophies characterized by short stature, a long, narrow thorax with short ribs, short limbs, variable polydactyly, and cystic renal disease. It is well known as a ciliary disease with multiple causative genes, many associated with intraflagellar transport in the cilium. Overlapping genes associated with JS include CEP120, CSPP1, KIAA0586, and PIBF1. In addition to JATD, skeletal features seen in JS are often described in related and overlapping skeletal dysplasias including Ellis-van Creveld syndrome, Mainzer-Saldino syndrome, Short rib-polydactyly syndrome, and Cranioectodermal dysplasia (Sensenbrenner syndrome) [4].
Leber Congenital Amaurosis (LCA, OMIM PS204000)
Leber Congenital Amaurosis (LCA, OMIM PS204000) is a severe form of congenital retinal blindness and nonrecordable electroretinograms. Genetically heterogeneous, with at least 19 genes and typically autosomal recessive but occasionally autosomal dominant inheritance [54]; about 25% of children also have cognitive impairment. The CEP290 gene has been implicated in 20% of LCA due to a cryptic intronic splice donor site as well as JS, MKS, and Senior-Løken syndrome [13, 54].
Meckel syndrome (MKS, OMIM PS249000)
Meckel syndrome (MKS, OMIM PS249000), also known as Meckel-Gruber syndrome, is an autosomal recessive disorder characterized by the triad of cystic dysplastic kidneys, brain malformations of the posterior fossa (usually occipital encephalocele), and the hepatic ductal plate malformation resulting in congenital hepatic fibrosis and proliferation of bile ducts. This condition is often lethal in the prenatal or perinatal periods [43]. At least 21 different genes have been implicated in Meckel syndrome, and for at least 18 of them, pathogenic changes have also been identified in JS (see Table 1). In some cases, more severe variants are associated with Meckel syndrome while variants predicted to have milder effects on protein function are associated with JS [83, 98]. There are also families in which the identical pathogenic variants in the same family can cause both Meckel syndrome and JS [82, 114].
Nephronophthisis (NPHP, OMIM PS256100)
Nephronophthisis (NPHP, OMIM PS256100) was one of the first ciliary disorders to be described, and is clinically manifest as polydipsia and polyuria due to impaired urinary concentrating ability, accompanied by chronic tubulointerstitial nephritis and later development of medullary cysts; it progresses to end-stage renal disease by age 30. About 80–85% of individuals have purely kidney disease, and ∼20% of subjects with juvenile nephronophthisis have a large, homozygous 290-kb deletion in the NPHP1 gene [44, 99]. A few individuals with JS have harbored this same deletion or other mutations in this gene [73]. NPHP is often associated with retinal dystrophy, a combination known as
Oral-facial-digital syndromes (OFD, multiple OMIM)
Oral-facial-digital syndromes (OFD, multiple OMIM) describe a number of disorders with characteristic facial features, oral abnormalities such as tongue lobulations, missing teeth, and extra oral frenula, and polydactyly. At least 13 clinical subtypes have been described, and the spectrum of findings overlaps considerably with MKS, short-rib thoracic dystrophies, and JS. In contrast to other forms of OFD which adhere to autosomal recessive inheritance,
Additional challenges in genetic diagnoses of ciliopathies
There are several major conundrums in understanding the genetic basis of ciliary disorders. One is the sheer number of genes that is causative for each clinically-defined disorder. Even a seemingly simplified ciliopathy such as nephronophthisis (NPHP) can be accounted for by over 20 genes, all of which are associated with the cilia, centrosome, or mitotic spindle [99]. In fact, most surveys report that about 15–20% of individuals with nephronophthisis are reported to have additional non-renal ciliopathy symptoms, the most common being RP. The NPHP1 gene only accounts for ∼20% of individuals with NPHP, with a homozygous deletion of the entire gene being the major mutation. Even in the situation where an individual harbors this clear biallelic loss-of-function deletion, however, the heterogeneity of the phenotype can range from NPHP to Senior-Løken to JS. Each of the remaining 20+ causative genes accounts for less than 1% of cases of NPHP, suggesting that over 60% of cases do not yet have a known genetic cause, in spite of 20 years of genetic studies on this condition [99]. It is notable that many ciliopathy conditions have multiple causative genes, with few individual genes causing more than 10–20% of cases within a specific condition. This lack of predictive ability and paucity of genotype-phenotype correlations are challenging.
The second conundrum is the extreme genetic heterogeneity of molecular causes and, in particular, the overlap of many causative genes with multiple different ciliopathy conditions. As noted, many of the genes that cause JS can also cause MKS, NPHP, OFD, BBS, and/or Jeune syndrome, or even more obscure ciliopathy conditions. When looking for genotype-phenotype correlations that might explain why the same gene can cause a lethal MKS phenotype as well as a milder JS phenotype and even more focal nephronophthisis, the severity of mutation has been implicated (e.g., 2 mutations in CC2D2A or TMEM67 resulting in frameshift or stop codons cause MKS while “milder” missense mutations cause JS) [8, 68]. Similarly, mutations in TMEM231 can cause MKS, OFD, and JS, with different presentations due to the same mutation even within the same family [61, 101]. In addition, different missense mutations in the same gene can cause disparate phenotypes, such as mutations in CEP290 that can cause JS, BBS, LCA, MKS, or Senior-Løken syndrome; it is likely that alterations in this protein, which straddles the MKS and NPHP modules within the transition zone, have different effects depending on their impact on interacting protein partners or different functional domains of the protein [90, 115]. Another possible explanation for this variability in the different outcomes for pathogenic variants in the same gene is that they may impact discrete isoforms of the protein that have distinctive functions. This is certainly the case for at least one BBS gene, ARL6, where one isoform is part of the BBSome and results in the BBS phenotype, whereas a mutation in a longer isoform has a specific role in photoreceptor maintenance [78]. Finally, it is likely that modifier genes or other epistatic effects such as differences in genetic background play a role. In these situations, proteins with overlapping functions and roles can be revealed. The specific manifestations of the pleiotropic effects of BBS may be partially explained by the unique roles of proteins associated with intraflagellar transport that underlie this condition: for example, ciliary biogenesis is dependent on Hedgehog signaling and is necessary for embryonic development, particularly of the nervous system and limb bud— consequently, mutations in many IFT components are associated with intellectual disability and skeletal dysplasias such as JATD [91]. Likewise, mutations in genes that impact the tissue-specific cargo of the IFT system may result in specific manifestations of ciliopathies [82]. The condition with the most overlap with JS is MKS, and both are associated with causal mutations in overlapping genes whose protein products localize to the transition zone. There is redundancy in the system, as the MKS, NPHP, and BBSome complexes likely have partially overlapping roles in promoting the ciliary localization of proteins and forming a barrier for protein transit into and out of the cilium. For proteins within the transition zone, a non-pathogenic variant may thus modify the phenotypes caused by pathogenic variants in a gene encoding a protein with overlapping or partially redundant function [82].
The third conundrum is the finding of occasional oligogenicity (variants in 2 or more different genes cause the condition; [46]), digenic inheritance, or triallelism in JS, as well as other ciliopathies. Triallelic inheritance refers to the observation that biallelic mutations in one gene are required along with a heterozygous pathogenic variant in a different ciliary gene to cause the disease [50]. Of course, these rare reports may reflect extremes of genetic modifiers, in which variants in other genes in addition to biallelic pathogenic variants in one known causative gene, affects the spectrum and severity of the phenotypic findings [75]. Several specific examples have been described, such as a heterozygous AHI1 mutation in combination with biallelic NPHP1 mutations resulting in a more severe brain phenotype in NPHP [111]. An NPHP1 mutation or deletion may also contribute to the severity of the BBS phenotype caused by 2 BBS biallelic mutations [59]. This has also been demonstrated with a common variant in RPGRIP1 L that modifies the retinal degeneration phenotype in a number of ciliary disorders [52]. Before the advent of next-generation sequencing capabilities, identifying genetic modifiers was challenging, and even targeted gene panels might fail to provide a comprehensive evaluation of all likely contributing modifying genes. One recent effort sought to tease out the prevalence of digenic inheritance, triallelism, and genetic modifiers in a large JS cohort [75]. Although rare disease variants were identified in over 1/3 of affected individuals in addition to the causal pathogenic mutations, they did not correlate with severity of the condition, nor did they support the presence of triallelic inheritance. Importantly, the rate of discordant phenotypes (∼60%) between affected siblings with shared pathogenic variants in a known JS gene suggested that modifying genes play an important role in JS, but they may be difficult to identify [75]. Another comprehensive study of the genetic contributions to a large cohort of subjects with ciliopathy phenotypes in a mostly consanguineous population demonstrated that ciliopathies are, for the most part, Mendelian disorders, as the burden of rare variants in known ciliopathy genes in the cohort did not differ significantly from the rate in a control cohort with non-JS intellectual disability; these authors suggested that stochastic events rather than modifier alleles contributed to phenotypic variability within their population [97].
Future directions in Joubert syndrome biology, genetics and therapeutics
The sheer number of causative genes for ciliary disorders such as JS can seem overwhelming, and clearly, identifying the causative gene and variant for a given family can be daunting. This is compounded by the lack of clear and consistent founder effects in isolated populations with these conditions. More research to identify additional causative genes will be critical for this effort, and new methodologies such as whole exome and whole genome sequencing are clearly paving the way, although interpretation of the meaning of variants can be quite challenging given the heterogeneity of genotypes and lack of correlation with phenotypes. Certainly, these features point to some level of redundancy in the complex biology of the cilium. And the extreme pleiotropy of organ system involvement in JS and other ciliopathies suggests that a deeper understanding of the roles of ciliary proteins, their interactions, and tissue specificity is necessary. In addition, the emerging role of modifying genes, many of which are ciliary genes as well, points to the role of ciliopathies serving as a bridge between strictly mendelian disorders and more complex polygenic disorders. For the extreme and rare cases of digenic or triallelic inheritance, the concept of threshold for mutational load appears operative. And in general, many of the principles that hold for one ciliary disorder are likely to be generalizable to other ciliopathies, suggesting that there may be some concepts of ciliary genetics and biology that are applicable to several, if not most, ciliopathies. For example, although many genes and several different signaling pathways are implicated in the pathogenesis of NPHP, the different mechanistic processes converge on the final common endpoints of renal tubular damage and fibrosis leading to ESRD [99].
In spite of lack of knowledge about the functional and developmental roles of many ciliarry proteins, there remains hope for development of therapies to impact specific ciliary phenotypes. Rescuing the developmental brainstem and cerebellar malformations that underlie the MTS may not be achievable, at least in the foreseeable future, but interventions to ameliorate the co-occurring morbidities such as retinal, renal, and/or liver disease could significantly impact quality of life for those with JS. One strategy to approach the cystic renal disease of NPHP has been to treat the elevated levels of cAMP and defects in urinary concentrating ability by use of vasopressin receptor-2 antagonists such as tolvaptan; animal models paved the way for the use of this drug in clinical practice and in current trials of childhood AD-PKD [69]. Other approaches to treat NPHP have included CDK inihibitors, SHH agonists, and mTOR pathway inhibitors such as rapamycin, but many of these are in early stages of development [99]. Gene therapy holds some promise in treating cystic renal disease in JS due to CEP290 mutations; an antisense oligonucleotide (ASO) has been used to promote alternative splicing and exon skipping to rescue an intronic mutation in this gene in patient-derived renal epithelial cells and in a mouse model of JS, with reduction of cystic kidney disease burden [81]. Similarly, a comparable approach to rescuing the retinal phenotype of LCA caused by the common intronic splicing defect in the CEP290 gene using gene therapy approaches has demonstrated some success in a cell model and in mice carrying the mini-gene [29]. Finally, as other articles in this special issue point out, there are novel strategies emerging to treat these and other manifestations of ciliopathy conditions (“Retinal disease in ciliopathies: recent advances with a focus on stem cell-based therapies review” by Chen, H et al., “Novel Treatments for Polycystic Kidney Disease” by Patil, A et al., and “Using human urine-derived renal epithelial cells to model kidney disease in inherited ciliopathies” by Sayer, J and Molinari, E.).
Footnotes
Acknowledgments
The author thanks the many families who have participated in past and current studies of Joubert syndrome and related ciliary disorders, the Joubert Syndrome and Related Disorders Foundation for their support, and many professional collaborators, including Drs. Meral Gunay-Aygun, Ian Glass, and Daniel Doherty.