
Abstract
Charcot-Marie-Tooth disease (CMT) is a progressive hereditary neuromuscular neuropathy with pathology in the myelin sheath or the axon. CMT caused by mutations in the Ganglioside-induced differentiation associated protein 1 (GDAP1) gene has been described by a spectrum of phenotypic presentations. GDAP1 is a mitochondrial protein responsible for protecting neuronal bodies from oxidative stress. It is associated with axonal and demyelinating pathophysiology with recessive and dominant modes of inheritance.We describe a case of a 9-year-old Puerto Rican female with clinical and electrodiagnostic results compatible with an axonal sensory-motor neuropathy where a genetic test describes a homozygous GDAP1 missense mutation at the c.692C>T (p.Pro231Leu), previously undetected in a pediatric Latino patient. Mutations in GDAP1 have been previously described in Tunisian, Old Order Amish, European and Japanese families with varying modes of inheritance. To our knowledge, this homozygous variant presentation of the GDAP1 gene is the first to be described in a pediatric Puerto Rican patient without a family history of hereditary sensory motor neuropathy.
Introduction
Charcot-Marie-Tooth disease (CMT) is one of the most common primary hereditary peripheral neuropathies with an estimated prevalence of 10–30 in 100,000 depending on country of origin [1]. CMT, otherwise known as Hereditary Sensory Motor Neuropathy, has two primary neuronal presentations: CMT1 and CMT2. CMT1 is a demyelinating subtype associated with reduced nerve conduction velocity and segmental demyelination/remyelination. Meanwhile, CMT2 is characterized by axonal loss without demyelinating lesions [2]. These are clinically characterized by progressive sensory loss, weakness, muscle atrophy, loss of deep tendon reflexes and foot deformities [1]. Basic criteria for CMT1/CMT2 in electrodiagnostic studies is a nerve conduction velocity of < 38 m/s in the motor fibers of the median nerve [2].Genetic heterogeneity in CMT ensures the same phenotype of hereditary peripheral neuropathy associated with different mutations of numerous genes.
Hereditary axonal neuropathies are caused by different mutations. The GDAP1 mutation is associated with an axonal or intermediate form of CMT, with recessive or dominant modes of inheritance and a wide range of severity [1]. This specific protein is involved in mitochondrial morphology and functioning. Mutations in the GDAP1 gene show a wide range of severity and Mendelian heterogeneity [7] leading to several forms of CMT including a recessive axonal form (AR-CMT2K). GDAP1 gene locus was first identified on chromosome 8q13-q21 (CMT4A) in Tunisian families and was associated with slow motor nerve conduction velocities and hypomyelination detected by nerve biopsy [1].
We describe a case of a 9-year-old female with a clinical presentation of a sensory motor peripheral axonal neuropathy with DNA testing remarkable for a GDAP1 variant of uncertain clinical significance. This case is novel in the Puerto Rican population.
Case presentation
A 9-year-old female arrived at the Pediatric Rehabilitation clinic with gait difficulty. She was independent in ambulation and activities of daily living with proper developmental milestones until approximately 2 years of age. At that time, she experienced frequent falls and limitations in mobility. In comparison to children of the same age, the patient was experiencing balance disturbances as well as difficulty engaging in running and jumping. Once she reached pre-school age, she presented impaired fine motor skills when compared to her peers. The parents deny any report of immediate or extended family history of similar presentation.
Physical examination at initial evaluation was remarkable for intact higher-level function, cranial nerves, facial symmetry, and normal phonation. Inspection of extremities was remarkable for mild bilateral pes cavus, without gross evidence of bilateral hand muscle atrophy. Sensory examination was unremarkable for vibration, pinprick, soft touch, and 2-point discrimination. Range of motion and strength testing were noticeable for impaired bilateral dorsiflexion. Deep tendon reflexes were +1 throughout except for 0 at bilateral Achilles. Ambulation was noticeable for bilateral foot drop and difficulty climbing stairs with the use of railings (Fig. 1).
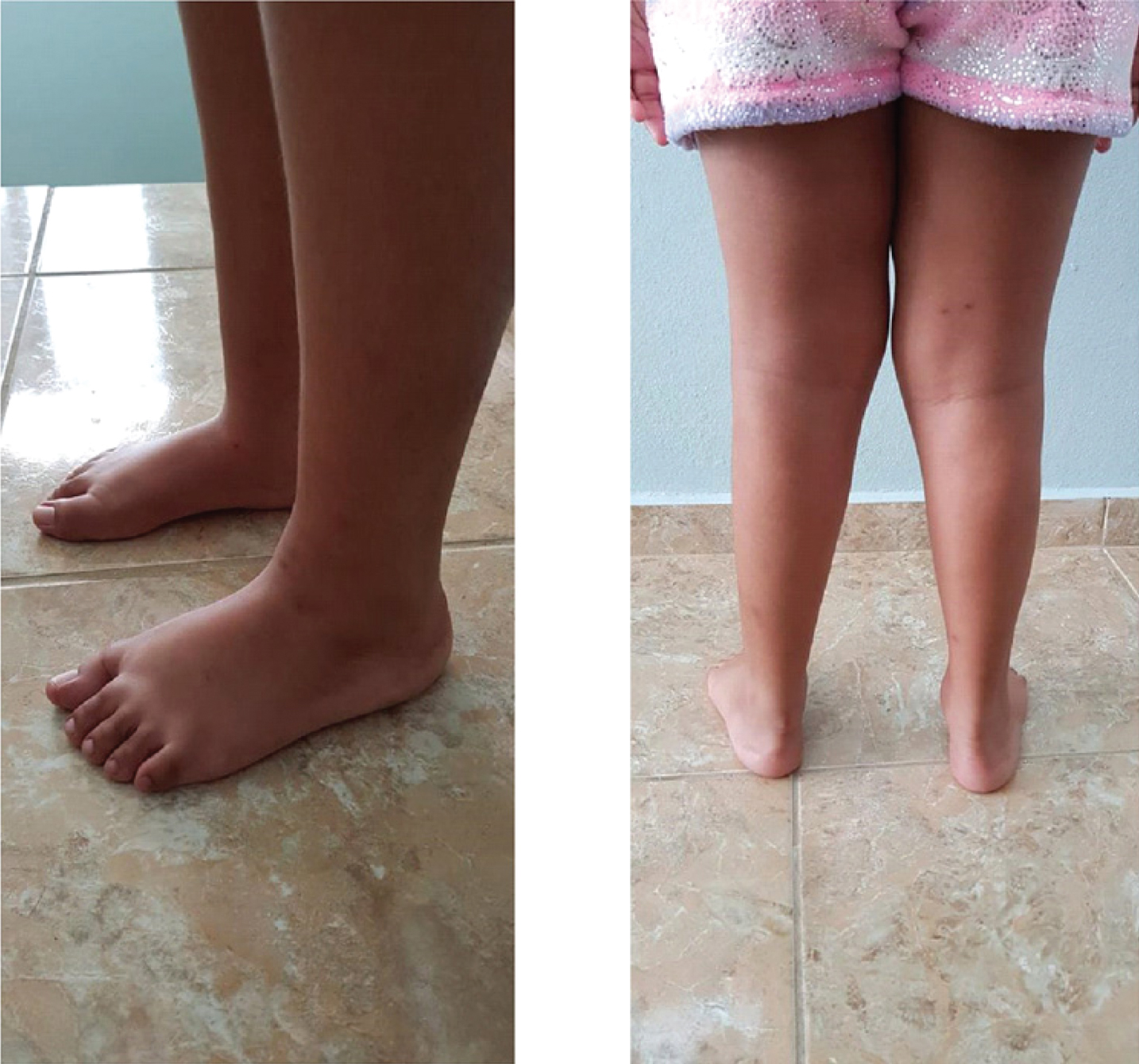
Lower extremity physical findings with mild bilateral pes cavus.
Ultrasonography of the median nerves was performed to assess cross sectional area (CSA) at the wrist and midarm. The right median nerve’s CSA was 12 mm2 and 14 mm2, at the wrist and midarm, respectively (average median nerve CSA 9–14 mm2) [3]. Electrodiagnostic studies were performed with evidence of an axonal sensory-motor neuropathy (see Table 1). Needle examination of lower limbs showed increased insertional activity with denervation potentials, normal motor unit potential parameters, and a neurogenic recruitment pattern of different myotomes (see Table 2).
Motor Conduction Studies
ms: milliseconds; mV: millivolts; mm: millimeters; m/s: meters per second.
Interference EMG
MUAP: Motor Unit Action Potential; EMG: electromyography.
Given the results, DNA testing using a hybridization-based protocol sequencing analysis and deletion/duplication testing for axonal neuropathy of 45 total genes were performed. Results were remarkable for a homozygous missense mutation, a variant of uncertain clinical significance in GDAP1 (p. Pro231Leu). The child’s parents were also tested, and results were remarkable for a heterozygous variant of the GDAP1 mutation gene without clinical manifestation of the neuropathy. As a result, a three-generation pedigree was obtained. Upon review, there were no relatives with similar clinical presentations (Fig. 2).
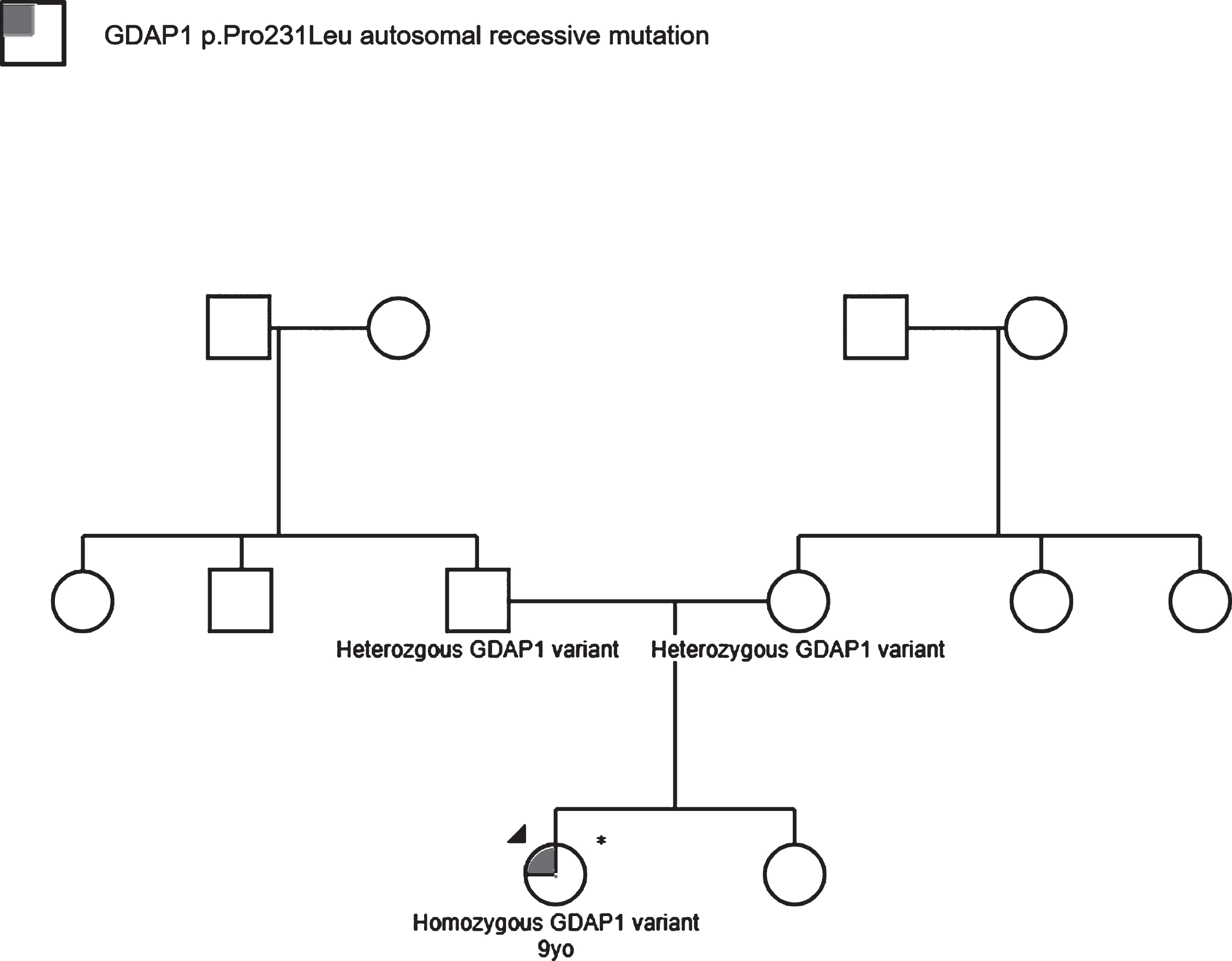
Family’s pedigree chart depicting an autosomal recessive inheritance of the GDAP1 genetic mutation.
GDAP1 gene mutations have been described as a cause of CMT subtypes. GDAP1 is associated with either the axonal or intermediate forms of CMT with recessive or dominant modes of inheritance and a wide range of severity phenotypes [2]. This locus was first identified in cases of recessive CMT in Tunisian families on chromosome 8q13-q21. This specific protein is involved in mitochondrial morphology and functioning. GDAP1 is a protein of the mitochondrial outer membrane which belongs to the glutathione S-transferase (GST) family of enzymes, known to catalyze the conjugation of the reduced form of glutathione with xenobiotic substrates resulting in intracellular detoxification [2] (see Fig. 3). This outer membrane protein is primarily expressed in nervous tissue, predominantly in axons within the peripheral nervous system. The expression in human skeletal muscle cells has also been evidenced with a direct relation to its metabolic regulation, glucose transport, and activation of mitochondrial biogenesis [4]. Yet, the function in other organ tissues is still unclear. In vivo studies indicate GDAP1 protects cells against oxidative stress [1]. In mouse neuronal cells, protein downregulation increases susceptibility against glutathione reduction [1]. This is of utmost importance, as neurons are polarized cells demanding a high level of energy that mitochondria can provide through ATP induced by oxidative phosphorylation [1]. Mutations in this gene are responsible for axonal damage primarily due to disrupted transfer of the mitochondria in the peripheral nerves which may account for axonal loss [1]. Depletion or variation of the gene leads to changes in the mitochondrial network [5].
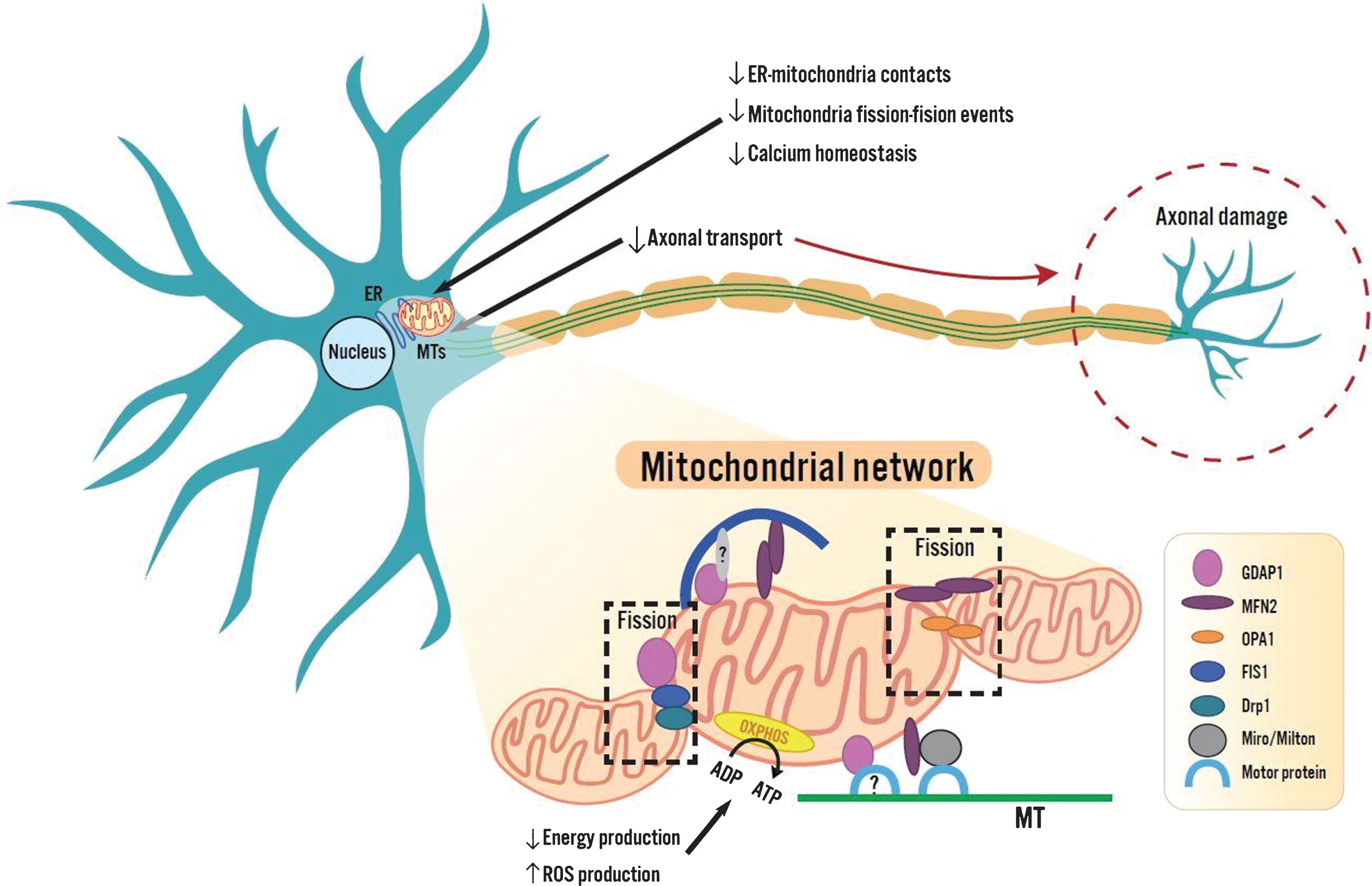
Mitochondrial network portraying the outer membrane proteins belonging to the glutathione S- transferase family of enzymes, catalyzing the conjugation of the reduced glutathione with xenobiotic substrates. GDAP1 is involved in multiple pathways (ER-mitochondria contacts, mitochondrial fission-fusion events, calcium homeostasis, and axonal transport), ultimately causing axonal damage.
The sequence change described replaces proline with leucine at the 231 codons of the GDAP1 gene resulting in a moderate physicochemical difference. Proline and leucine are both non-polar hydrophobic amino acids belonging to the same group. There is still no evidence describing the potential pathogenic changes that this missense substitution may imply. However, given their primary structure characteristics, it can be hypothesized that this substitution can alter the secondary protein structure the gene encodes given our patient’s clinical presentation.
To date, more than 80 variants of this gene, both missense and nonsense mutations, resulting in possible structure and function alteration of proteins have been described [7]. Among the recessively inherited changes, nonsense and frameshift mutations have been associated with severe phenotypes of CMT [5]. CMT involving the GDAP1 mutations generally leads to aggressive disorders appearing in infancy or early childhood [6]. In most cases, the disease leads to disability such as wheelchair dependency before the second to third decade of life. In some cases, the presence of vocal cord paresis and diaphragmatic paralysis suggests the clinical progression of the disease [7]. Cassereau et al. (2011) evidenced that the recessive forms of CMT disease involving GDAP1 mutations are far more severe than the dominant forms. The dominant forms present with a milder adult-onset phenotype with distal involvement and slow progression [7]. Autosomal recessive forms of CMT are less common in the general population but account for the vast majority of CMT phenotypes in communities with a high prevalence of consanguinity. In this case, this was not present. Pakhrin et al. (2018) found the c.692C > T (p.Pro231Leu) variant in exon 5 of the GDAP1 gene to be the most frequent pathogenic variant in the Old Order Amish population [8].
The authors were unable to find reports of this mutation in the pediatric Latino population. Vivar et al. (2019) published a case of a 33-year-old Puerto Rican male with distal weakness, atrophy, and DNA positive testing for a homozygous mutation c.692 > T (pPro231Leu) in GDAP1. Due to accessibility and lower cost of these genetics tests, we can expect that mutations in the GDAP1 and other genes will be more frequently described.
In conclusion, a case of childhood onset axonal CMT with a homozygous GDAP1 pathogenic variant was presented. The CMT variant has yet to be reported in the Puerto Rican/Latino population. The GDAP1 mutation must be considered as probable cause in the case of a pediatric patient evaluation with clinical and electrophysiological evidence of an axonal sensorimotor neuropathy.
Footnotes
Acknowledgments
Conflict of interest
The authors certify they have no affiliations with or involvement in any organization or entity with any financial interest or non-financial interest the subject matter or materials discussed in this manuscript.