
Abstract
The innate immune system of the brain is mainly composed of microglial cells, which play a key role in the maintenance of synapses and the protection of neurons against noxious agents or lesions owing to their phagocytic activity. In the healthy brain, microglia are highly motile and strongly interact with neurons either by physical contact, induction of oxidative stress or through specific mediators, such as chemokines and cytokines. In response to inflammatory insult however, microglial cells get activated and produce inflammatory cytokines. The action of cytokines on specific receptors expressed in the brain triggers the development of sickness behavior and altered cognitive and emotional processes. The effects are acute and reversible as normal behavior is restored once the synthesis of inflammatory brain cytokines returns to baseline after a few hours. However, in pathological situations, these cytokines may reach toxic levels and have irreversible consequences such as neuronal death, as observed in neurodegenerative disorders such as Alzheimer's disease. Omega-3 (n-3) polyunsaturated fatty acids (PUFAs) are essential nutrients and fundamental components of neuronal and glial cell membranes. They accumulate in the brain during the perinatal period in a dietary supply-dependent fashion. Their brain levels may diminish with age, but can be increased by diets enriched in n-3 PUFAs. Changes in the immune profile have been associated with n-3 PUFAs intake in humans and animal models. Therefore, the increasing exposure of the population to diets low in n-3 PUFAs could contribute to the deleterious effects of the chronic activation of microglia in the brain.
Introduction
The central nervous system (CNS) has long been considered as an immunoprivileged organ. Indeed, in physiological conditions, the blood brain barrier (BBB), owing to its tight junctions, considerably restricts the entry of immune cells, notably lymphocytes, into the brain. Research in neuroimmunology has shown that the brain possesses its own line of defense, activated by immune stimuli, and closely linked to the peripheral immune system. Although not as isolated as first thought, there are important distinctions between the peripheral and the central immune systems. Inflammatory cytokines, which are important mediators of the communication within the immune system, also act in the brain where they can activate microglial cells and astrocytes that in turn, can produce cytokines, chemokines, complement proteins and nitric oxide [1–3]. In physiological conditions, the synthesis of brain immune mediators is finely regulated, allowing a rapid return to basal levels, without leading either to BBB failure or cerebral lesion. However, when synthesized in large amounts or in a chronic fashion, these factors may have toxic effects on neurons, resulting in substantial neuronal dysfunction that can lead to cell death [4–7]. During aging, chronic microglial reactivity and production of low levels of inflammatory cytokines occurs and can produce alterations of neuronal functions [8]. Indeed, this age-associated inflammation, characterized by the increased production of brain cytokines, increases the vulnerability of the aging brain to immune stimuli, and the risk of developing delirium and/or neurodegenerative disorders with an inflammatory component, such as Alzheimer's disease (AD) [9–11]. Accordingly, clinical and epidemiological studies have suggested a possible association between the systemic expression levels of inflammatory cytokines and the incidence of functional/behavioral alterations (cognitive or mood disorders) in psychiatric and elderly subjects [12–15]. In this context, limiting the development of chronic neuroinflammation could protect the brain against neurodegenerative disorders. This could be achieved with the diet, a modifiable environmental factor to which each individual is exposed throughout his life.
Increasing attention has been paid to omega-3 (n-3) and omega-6 (n-6) polyunsaturated fatty acids (PUFAs). These micronutrients are essential since their precursors cannot be synthesized de novo by the organism. Moreover, their synthesis is extremely limited in most mammals (less than 5% of the precursors are converted) [16]. Increasing data in animal models as well as in humans suggest that dietary PUFA exert immunomodulatory effects [17–20]. Indeed, n-3 long chain PUFAs form the basis of lipid derivatives (neuroprotectins and resolvins) with anti-inflammatory properties [21–26]. Moreover, they are precursors of eicosanoids including leukotrienes, prostaglandins and thromboxanes, which are important modulators of the inflammatory response. However, they have lower biological potency than eicosanoids derived from n-6 PUFAs such as arachidonic acid [27, 28]. The brain is extremely rich in PUFAs, and the accumulation of PUFAs in brain tissues first takes place during the perinatal period in proportions which are dependent on maternal dietary levels [29, 30]. Conversely, the levels of brain PUFA has been reported to diminish with aging, although this decrease can be prevented or corrected by appropriate nutritional strategies [31]. Since the Industrial Revolution, we have observed a decrease in energy expenditure related mainly to sedentary lifestyle combined with an increase in the consumption of high energy foods characterized by elevated levels of sugar, saturated fats, and n-6 PUFAs, poor in n-3 PUFAs, vitamins, proteins and micronutriments [32, 33]. The resulting imbalance in the n-6:n-3 ratio from this dramatic modification in the dietary intake is currently estimated to 10:1 to 20:1 in Western diets, whereas the current recommended ratio is 1:1 to 2:1 [32, 33]. With the known immunomodulation resulting from n-3 PUFAs intake, this imbalance could therefore contribute to prime the brain to the deleterious effects of inflammatory cytokines, and eventually to the development of neurodegenerative and/or neurobehavioral disorders.
The brain innate immune system (BIIS)
In the periphery, tissue injuries caused by trauma or pathogens induce a rapid local inflammatory response involving local cells and characterized by the synthesis and release of proinflammatory factors, among which cytokines and chemokines, followed by systemic recruitment of immune cells. The purpose of this local response is to eliminate pathogens and to promote tissue repair. However, the failure of resolving the insult and dysregulated injury may lead to chronic inflammation, which is toxic for the tissue, ultimately resulting in cell death. In addition, peripheral inflammation can also influence brain immunity [1]. However, it appears that the steps involved in the cerebral immune response are distinct from those of the periphery. Therefore, the term “neuroinflammation” is broadly used to discriminate the brain from the peripheral inflammation. “Neuroinflammation” describes the brain inflammatory response involving not only peripheral immune cells influx into the brain but also the specialized response of brain innate immune system (BIIS) [34].
Microglial cells are the main cells of the BIIS. They are the parenchymal resident macrophages of the brain where they act as a first line of defense(phagocytosis, antigen presentation, cell recruitment and secretion of cytokines) [35, 36]. They account for 5 to 20% of the non-neuronal glial cells, depending on the brain structures analyzed. Microglia are distinct from the brain macrophages found in the meninges, choroid plexus, and perivascular space, owing to their different developmental origin. Indeed, recent data highlight that microglia derives from macrophages produced by primitive hematopoiesis in the yolk sac [37–40] while brain macrophages derive from myeloid precursor in the bone marrow [41, 42]. Microglia precursors colonize the CNS during the embryonic and fetal phases of development [43]. Interestingly, an increase of CD11b+/F4/80+ microglia occurs in the post-natal brain of rodents [37]. However, recent evidence suggests that this increase in microglial cell number is not induced by the recruitment of blood-derived myeloid precursors but instead results from the proliferation of resident microglial cells [39, 44]. Recent studies provide new insights into the development of the microglial population from yolk sac progenitors during embryogenesis. In contrast with macrophages from other tissues, microglia persist throughout the entire life of an individual [42]. There is a growing interest to characterize microglia cells profile and signature. Very recent data reveals that microglia express a decreased number of mRNA types as compared to tissue macrophages [45] and display a specific mRNA signature that is dependent on TGFβ [46].
Microglia are particularly sensitive to changes in their microenvironment and readily become activated in response to infection, trauma or disease [47]. In healthy brain, the microglia can phagocyte apoptotic neurons and debris and reduce neuroinflammation which, in turn is beneficial to viable neurons [48]. However, in inflammatory or pathologic situations, the phagocytic adaptor protein MFG-E8 is released by microglia and binds to phosphatidylserine (PS) exposed on apoptotic neurons. This activates neuronal phagocytosis by microglia via the vitronectin receptor [49]. Annexin A1, another eat-me signal released by microglia, serves as a bridge with PS on dying neuron, helping the microglia to discriminate between apoptotic neurons and healthy neurons [50]. Recently, new data revealed that activated microglia in inflammatory state appears to lose their ability to discriminate between apoptotic and viable neurons, resulting in phagocytosis of diseased as well as healthy neurons [51].
In the adult brain, microglial cells have a ramified morphology when quiescent, and ameboid morphology when activated. Ramified microglia generally display less phagocytic activity and weakly express ligands and receptors involved in macrophage function. Disseminated throughout the brain parenchyma, they use their processes to receive signals such as danger-associated molecular patterns (DAMP) and pathogen-associated molecular patterns (PAMP) from their microenvironment, which reveal the existence of an endogenous pathological signal or the presence of a pathogen, respectively. In order to do this, microglial cells express a set of pattern recognition receptors (PRRs) including the Toll-like receptors (TLRs) that allow the recognition of PAMPs, such as the bacterial endotoxin [52, 53], and DAMPS, such as misfolded proteins [54], and promote brain inflammatory reaction [55]. The activation of PRRs by PAMPS and DAMPs induces a signaling cascade leading to the secretion of cytokines and chemokines. The microglial cells further coordinate the inflammatory response via the expression of membrane receptors for inflammatory cytokines interleukin (IL)-1β, tumor necrosis factor (TNF)α and IL-6 and several chemokines. In vivo, IL-1β, TNFα and IL-6 are produced by microglia in response to peripheral immune stimuli such as bacterial endotoxin lipopolysaccharide (LPS) [56].
The BIIS response promotes clearance of pathogens, toxic cellular debris and apoptotic cells and therefore protects the brain. Indeed, a selective ablation of proliferating microglia exacerbates brain damage in adult and in neonatal hypoxic ischemic injury models [57]. However, under some circumstances, the sustained expression of inflammatory factors such as cytokines can lead to neurodegeneration [7, 58]. The BIIS response is therefore a double-edged sword depending on a fine balance between protective and detrimental effects that needs to be tightly controlled. Real-time in vivo imaging technology has revealed that both in health and disease, microglia exist in an active state but their role in the brain depends on the broad phenotype spectrum they can adopt [59]. Microglia phenotypes, so called polarization, could be crucial in the protective or detrimental role of PRR-activated BIIS response toward neurons. According to what was described for macrophages, it has been suggested that activated M1 cells have cytotoxic properties, M2a are involved in repair and regeneration, M2b promote immunomodulation, while M2c have an acquired-deactivating phenotype [60] (Fig. 1). In vivo, microglia express proinflammatory cytokines associated with a M1 phenotype (IL-1, IL-6, IL-12 and TNFα) in response to an immune stimulus. Recent evidence indicates that neurons exert some control on microglia activity [61]. The extent of neuroinflammation therefore depends on the bi-directional interactions between neurons and microglia. Recruitment and activation of microglial cells require well-organized reciprocal communication between these two cell types [36, 62]. As a result, neurons release ON or OFF signals to regulate the activation of microglia. On the one hand, OFF signals (CD200, CX3CL1, CD47, CD55 and HMGB1) are produced by healthy neurons to keep microglia in their surveillance mode. On the other hand, damaged neurons express inducible ON signals (chemokines, purine and glutamate) to activate microglia and phagocytosis [36]. Interestingly, such neuron-glia interaction is impaired in the aged brain leading to amplified and prolonged microglial activation and production of proinflammatory cytokines [63, 64].

Microglia phenotype plasticity. Microglia can adopt different phenotypes: M1 (classical activation), M2a (alternative activation), M2b (immunoregulatory) and M2c (acquired-deactivation). According to their phenotype, microglia cells express different clusters of differentiation (CD) such as CD86 or CD206, or type-II proteins of major histocompatibility complex (MHC) and secrete different cytokines and chemokines. CCL: chemokine (C-C motif) ligand; IFN: interferon; IL: interleukine; LPS: lipopolysaccharide; TGF: transforming growth factor; TNF: tumor necrosis factor; Ym1: YKL-40, chitinase 3-like 3.
Neuroinflammation, the aged brain and Alzheimer's disease
Aging is associated with senescence of microglia, impaired microglia phagocytic activity and low-grade neuroinflammation [65], which is characterized by a higher expression of proinflammatory cytokines IL-1β, IL-6 and TNFα to the detriment of anti-inflammatory factors such as IL-10 and IL-4. This state is called inflammaging at the periphery and in the brain [66]. The overproduction of proinflammatory cytokines in the absence of infection or injury in the aged brain could be linked to the impairment of microglial activity. Indeed, microglia number and activity increases during normal aging [67]. These aged cells, in addition to producing proinflammatory cytokines, contain lipofuscine granules, and decreased processes complexity, a morphological change found in activated microglia [47, 68]. In addition, microglia in the aged brain express higher levels of CD86, major histocompatibility complex II (MHC II), TLR and CR3/CD11b which are markers of activation [69, 70]. Senescent microglia display reduced phagocytic activities of beta-amyloid in aged transgenic mice which could be due to their M1 phenotype [71]. Mechanisms involved in increased microglia activation in the aged brain are not fully understood, however as CD200 and CX3CR1 expression is impaired it could be that neuron-glia interactions are disturbed [67, 72].
The involvement of microglia in the communication of systemic inflammatory signals to neurons has been recently reviewed [73]. One of the most important new knowledge is that microglia become more susceptible to inflammatory stimuli when they are primed by pathological insults contributing to the progression of neurodegenerative diseases such as Alzheimer's Disease (AD) [73, 74]. The characteristics of primed microglia remain to be determined. However, increased number of microglia, together with changes in morphology and increased expression of cell surface antigens such as CD68, complement receptors (CR3) and/or MHC have been consistently reported in priming context as first reported in prion disease [75]. Priming phenomenon has been observed in aging [76], after the administration of proinflammatory endotoxins [77, 78] and in animal models of AD and Parkinson disease [7, 79]. Interestingly, aging microglia express a specific sensome (defined as proteins sensing microbes) that could confer them a higher vulnerability to inflammatory stimuli [70]. Indeed, the concept of microglia priming provides new view of how systemic inflammation could contribute to the progression of neurodegenerative diseases, through increased production of inflammatory cytokines that could damage neurons and/or loss of neuroprotective properties.
The first evidence of an inflammatory response in AD comes from case-control studies showing immune anomalies in blood or CSF from living individuals or cerebral tissue collected post-mortem. For example, plasma cytokine profiles, key components of the complement system and immune cells are altered early in the disease [80–83]. High levels of pro and anti-inflammatory factors, and increased PRR and chemokines expression are found in the brain of AD patients [4, 84]. Activated microglia in the brain of AD patients have been detected by (R)-[3H]PK11195 PET binding to peripheral benzodiazepine receptors [85]. A second set of evidence comes from epidemiology. Indeed, chronic non-steroidal anti-inflammatory drugs (NSAID) use has been associated to lower risk of AD [86, 87]. Finally, data generated in animal models suggest a causal link between brain amyloid or tau pathologies and the triggering of an immune response [5, 89], although this view is also disputed [90]. In transgenic APP/PS1 or 3xTg-AD mice, reports suggest that microglia activation starts between 6 and 12 months, as assessed with CD45, Iba1 and/or F4/80 IHC [91–94]. Therefore, despite conflicting data, it appears clear that changes in parameters involved in central or peripheral inflammation are associated with AD.
However, the role of inflammation or the immune system in AD pathogenesis is still a matter of debate as both beneficial and adverse effects of inflammation have been reported [95–101]. Recent data highlighted that microglia, because of its impaired activity in the AD brain, cannot phagocytocize Aβ that therefore accumulates [102, 103]. In turn, Aβ accumulation activates microglia in a chronic proinflammatory state that contributes to the disease progression and, ultimately cognitive decline [4]. Higher levels of IL-1 have been implicated in both the initiation and progression of neuropathological changes [104]. Accordingly, overexpression of IL-1 in the AD brain has been linked to an increased microglial activity, frequently associated with amyloid plaques [105]. In addition, brain from Tg2576 mice (a model of AD) exhibits significant increases in IL-1 expression in comparison to healthy animals [106]. Overexpresssion of IL-1β in the 3xTg-AD mouse model of AD suggest that amyloid and tau pathology are differentially regulated, with a reduction in amyloid deposit but an exacerbation of Tau hyperphosphorylation [107]. Such effects could involve the fractalkine pathway as the expression of its receptor CX3CR1 have been reported to be associated with either a decrease [108] or an activation of Aβ clearance [109–112] and increase Tau phosphorylation [113]. Therefore, further studies are needed to precise the role of the fractalkine pathway in these processes.Globally, a crucial issue that remains unclear is whether the immune response observed in AD is a compensatory mechanism with a neuroprotective effect, part of a vicious circle leading to neuronal death and cognitive decline, or both [72, 115]. Answers to this question will determine how neuroinflammation can be “tamed” for the purpose of developing therapeutic interventions.
In the last decades, trials aiming at modulating brain inflammation in AD patients have been dis- appointing [72, 116–118]. This included assays with NSAID, steroid anti-inflammatory (prednisone) or with immunomodulators such as TNF antagonists and polyclonal or monoclonal immunoglobulins [72, 119]. However, consistent with epidemiology data [86], it remains likely that the modulation of inflammation is more suitable for a preventive intervention [120]. Unfortunately, no such preventive study has been completed yet, the ADAPT study on COX-1 and COX-2 inhibitors having been ended abruptly due to unsuspected side effects [72, 121]. Nevertheless, further data analysis from the ADAPT study do not provide a clear answer to the hypothesis that celecoxib or naproxen exert a preventive action against AD [122, 123]. Because the role of BIIS in the development of AD is complex, other strategies aiming at optimizing microglia activity, rather than just blocking inflammatory factors synthesis in the brain could be more beneficial [4, 72].
Polyunsaturated fatty acids and neuroinflammation
N-3 PUFAs: A brief overview
PUFAs of the n-3 or n-6 families are essential nutrients, as the precursors of these two series (linoleic acid (18:2n-6, LA) and α-linolenic acid (18:3n-3, ALA)) cannot be generated de novo in mammals, they have to be provided by the diet. They are respectively metabolized by a series of elongation and desaturation steps into arachidonic acid (20:4 n-6, AA) and eicosapentaenoic acid (20:5 n-3, EPA) and docosahexaenoic acid (22:6 n-3, DHA) (Fig. 2). These long chain PUFAs are incorporated into cell membranes as phospholipids. The liver is the main site of conversion of LA and ALA into long chain PUFAs, although other organs such as the brain also express the necessary elongases and desaturases [124]. Since the two series of PUFAs compete for the use of the enzymes necessary for their biosynthesis, and because the conversion into long chain is extremely limited (less than 5% of the precursors is converted), their supply by the diet is of particular importance. Foods that were previously consumed by humans were obtained from hunting and fishing and were relatively rich in n-3 long chain PUFAs. Since the Industrial Revolution, the ratio of n-6:n-3 PUFAs in the diet has increased from 1 to almost 20 in industrialized countries like the United States, leading to a suboptimal dietary consumption of n-3 PUFAs [33, 125].
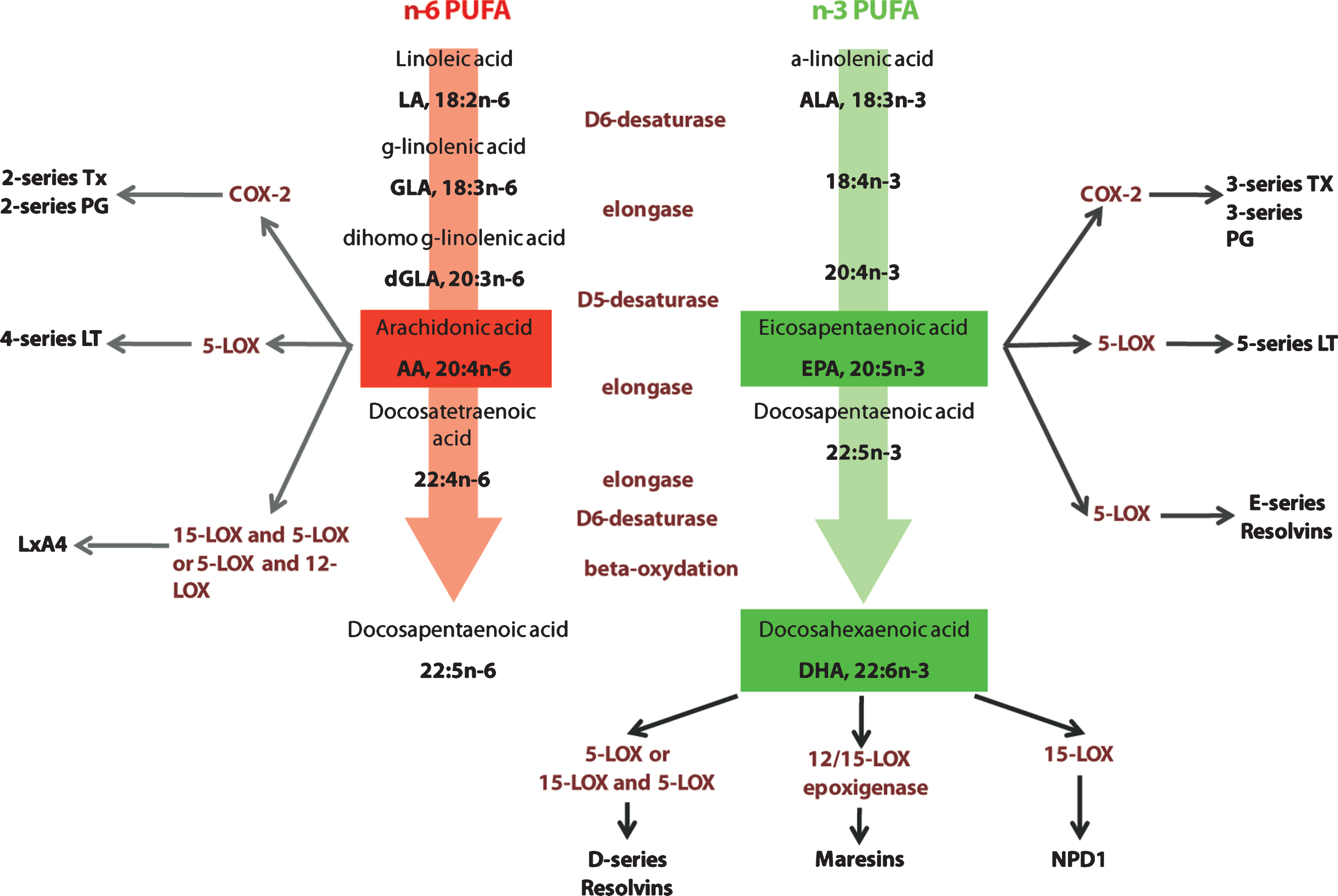
Elongation and desaturation of n-6 and n-3 PUFA and pathways in active eicosanoid metabolism from arachidonic acid, eicosapentaenoic acid and docosahexaenoic acid. n-6 and n-3 essential fatty acids precursors are linoleic acid (LA) and α-linolenic acid (ALA). These precursors are metabolized into arachidonic acid (AA) and eicosapentaenoic (EPA) and docosahexaenoic acid (DHA) respectively. AA is metabolized into derivatives that belong to the eicosanoid family, series 2 and 4. EPA and DHA metabolic derivatives belong to the eicosanoid family, series 3 and 5, resolving family (series D and E) and neuroprotectins. Tx: thromboxane; PG: prostaglandines; COX-2: cycloxygenase 2; LT: leukotrienes; LOX: lipoxygenase; Lx: lipoxin; NPD1: neuroprotectin D1.
The dietary deficiency in n-3 PUFAs is associated with a significant decrease in DHA in the brain, and could thus promote neuroinflammatory processes and the subsequent development of inflammation-related CNS disorders [8]. However, the effect of n-3 long chain PUFA supplementation to generate a favorable inflammatory marker profile is currently subject to debate. In populations with a high n-3 long chain PUFA dietary intake due to elevated fish consumption, such as Greenland Inuits, the incidence of ischemic heart and autoimmune diseases (such as psoriasis, asthma or multiple sclerosis) is low [126, 127], but inflammatory markers such as YKL-40 and hsCRP are rather found to be increased [128]. While some clinical studies have reported anti-inflammatory effects of n-3 long chain PUFAs administered in the context of chronic and autoimmune inflammatory disorders, other reports fail to reproduce these findings (for review [28]).
Attenuation of the inflammatory response is one of the most frequently cited mechanisms of action of n-3 PUFAs, despite limited convincing evidence of such an action in cerebral tissues until recently [129]. As stated above, a high n-3 PUFA intake decreases AA content and at the same time, increases DHA in the brain. Since n-3 PUFAs compete with AA as substrates for cyclooxygenase (COX) and lipoxygenases (LOX), increased n-3 PUFA concentration is expected to reduce the production of the more potent inflammatory eicosanoids derived from AA [25, 130–132]. Therefore, food rich in n-3 PUFA such as fish or walnut have been found to limit the inflammatory response in preclinical studies [133–136].
More specifically, n-3 PUFAs also decrease the production of various important inflammatory cytokines such as TNFα, IL-1, and IL-6 [20, 137]. Indeed, DHA decreases the expression of brain inflammatory markers following systemic LPS administration [138], brain ischemia-reperfusion [20, 139] and spinal cord injury [140]. However it remains difficult to gauge the direct effect of DHA on BIIS, since the primary CNS injury is also attenuated in these studies. High-fat intake in n-3 PUFA-deprived animals induces a rise of GFAP, a marker of astrogliosis, in mouse brains [141]. Recently, we have demonstrated thatin vitro, in murine microglia induced by LPS, the production of IL-1β and TNFα by is strongly inhibited by DHA through its effect on LPS signaling pathway nuclear factor-κB [137]. In vivo, chronic dietary n-3 PUFA deficiency significantly increases the production and release of IL-6 and TNFα in the blood [142]. In addition, mice exposed throughout life to a diet devoid of n-3 PUFAs display lower brain DHA and higher LPS-induced IL-6 levels in the plasma and the hippocampus [138]. In parallel, n-3 PUFAs have been shown to decrease the levels of COX-2 in vitro [143] and in vivo [20, 144].
The recent discovery of a novel family of endogenously generated autacoids, namely resolvins and protectins, with potent anti-inflammatory and proresolving activities offer a better understanding of the mechanisms responsible for the protective effect of DHA in the brain [25, 145]. In particular, resolvin D1 (RvD1), which originates from DHA via lipoxygenases, promotes the resolution of inflammation and has been detected in the brain [21]. Very interestingly, DHA and RvD1 promotes macrophage polarization toward a M2 state in obese mice adipose tissue associated to a decrease of pro-inflammatory cytokines and an increase of anti-inflammatory cytokines [146, 147]. In a model of LPS injection, Orr et al (2013) [129] showed that DHA exerts its anti-inflammatory effects in the brain, via its conversion into resolvins. Neuroprotectin D1 (NPD1), a DHA derived docosanoid has also been detected in the brain where it could exert anti-inflammatory and protective activities [23]. Chronic infusions of DHA or NPD1 in the brain significantly decreased neuroinflammatory processes triggered by a middle cerebral artery occlusion [139]. NPD1 even had a more potent effect than DHA [139, 148]. However, it remains to be demonstrated that NPD1 is the intermediary of the anti-inflammatory effect of DHA in the brain.
Experiments conducted in animal models have highlighted brain DHA as a potent mediator of the protective effects of dietary n-3 PUFAs. Low dietary intake of n-3 PUFA decreases DHA levels in the animal brain [149–151]. As a result, emotional behavior (depressive-like symptoms and anxiety) as well as learning and memory are impaired as shown by others and us [152–155]. On the other hand, positive effects of diets enriched in DHA on learning and memory have been demonstrated in laboratory animals [156–160].
N-3 PUFAs and age-related neuroinflammation
As previously mentioned, PUFAs represent potent immunomodulatory agents. During aging, the levels and the turn-over rate of brain PUFAs decrease, particularly in the hippocampus, cortex, striatum and hypothalamus [161–164]. Brain levels of DHA and AA diminish in aging rats with alterations in cognition and in long-term potentiation (LTP) in the hippocampus [162]. In senescence-accelerated mouse (SAMP8), a spontaneous model of accelerated aging, DHA levels decrease, whereas lipid peroxidation increases with age including that of DHA [165, 166]. In addition, the conversion of the precursors LA and ALA into their long chain derivatives becomes less efficient. The activity of the Δ6 desaturase decreases with age in the liver and the brain [167, 168]. Phospholipid synthesis pathways are also altered with age, thus reducing the incorporation of PUFAs into membranes [169]. The combination and interaction of these different alterations associated with aging contributes to a reduction in the level of DHA, i.e. a reduction in the index of membrane fluidity, in the brain of elderly people. In animals, aging was found to be associated with a decrease in the membrane content of AA in the hippocampus together with an attenuation of LTP that can be reestablished by a diet containing AA [170].
With aging, IL-6 expression is increased in the cortex of both n-3 deficient and n-3 adequate CD1 mice while IL-10 expression is decreased with no effect of long term ALA deficient or enriched diet [155]. Conversely, short-term exposure to dietary EPA reduces IL-1-induced spatial memory deficit and anxiolytic behavior [171, 172] and improves LPS and Aβ-induced inhibition of LTP in both adult and aged rats [173]. The expression of markers of microglial activation (CD68, MHCII and CD11b) increases with age in animals, as does the number of microglia in the brain of humans, attesting of the occurrence of age-related neuroinflammation [174]. Microglial cell reactivity is involved in the age-dependent increase in the production of inflammatory cytokines, as demonstrated by the inhibition of inflammatory cytokine overexpression by minocycline in aged rats [69]. In rats, the age-related activation of the microglia, production of IL-1β and alterations in hippocampal LTP are attenuated by EPA [175, 176]. Importantly, a 2-month fish-oil dietary supply increases DHA in the brain, prevents proinflammatory cytokine expression and astrocyte morphology changes in the hippocampus and restored spatial memory deficits and c-Fos-associated activation in the hippocampus of aged mice [31]. These data support the idea of the importance of DHA dietary supply in aged mammals.
To detect an anti-inflammatory action in neurodegenerative diseases, one must work with an animal model displaying an easily quantifiable inflammatory response, which is not always the case in AD models [90]. In contrast, animal model of stroke offers an easier opportunity to directly probe this mechanism of action. To visualize the effects of DHA on stroke-induced neuroinflammation, we recently used the TLR2-fluc-GFP transgenic mice exposed to either (i) a control diet; (ii) a diet depleted in n-3 polyunsaturated fatty acid (PUFA) (iii) a diet enriched in n-3 long chain PUFA (0.7 g kg−1 day−1, with DHA:EPA ratio of 4:1) during 3 months [20]. Real-time biophotonic/bioluminescence imaging of the TLR2 response was performed before and after middle cerebral artery occlusion (MCAO), whereas cytokines concentrations and stroke area analyses were performed 3 and 7 days after MCAO, respectively. We have observed that 3 months of DHA/EPA treatment abolished the TLR2 response after ischemic injury, while increasing the brain n-3:n-6 PUFA ratio, preventing microglial activation, reducing the ischemic lesion size and increasing concentrations of the anti-apoptotic molecule Bcl-2 in the brain [20]. Additional analysis further revealed a significant decrease in the levels of COX-2 and IL-1beta, but not other pro-inflammatory cytokines [20]. The result of this study argues for the use of n-3 long chain PUFA in preventing the initiation of TLR2-dependent signaling cascade, which lays upstream of the main pathways leading to a neuroinflammatory response.
In epidemiological and observational studies, a higher level of blood n-3 PUFAs is associated with lower proinflammatory cytokine production [177–180]. In a cohort of elderly subjects, depressive individuals with an elevated plasma n-6:n-3 ratio were found to exhibit higher levels of TNFα and of IL-6 [180]. F2-isoprostane, a marker of oxidative stress, and telomere length, an indicator of immune cell aging, are both decreased in the blood of subjects supplemented with EPA/DHA [181]. Additionally, n-3 PUFA supplementation in elderly subjects reduced the levels of inflammatory cytokines produced by blood leukocytes stimulated in vitro [182]. The production of prostaglandin E2 by monocytes is inversely correlated to the EPA content of leukocytes obtained from aged subjects after the consumption of dietary complements containing different doses of EPA [183]. To the extent that the level of peripheral cytokines can reflect that of brain cytokines, these results would suggest that dietary n-3 PUFAs modulate neuroinflammation and associated neurobehavioral effects in elderly individuals [8] (Fig. 3).

Potential role of n-3 PUFA in inflammaging. In the aged or diseased brain (AD neuropathology), microglia are primed and polarized into various phenotypes (e.g. M1 in aging) and secrete pro-inflammatory cytokines that could play a role in cognitive impairment. The protective effect of n-3 PUFAs toward cognitive deficit in aging or neurodegeneration could be linked to the promotion of an anti-inflammatory M2 phenotype.
Epidemiological studies further highlight the importance of n-3 PUFA levels in the development of age-linked neurodegenerative disorders. Indeed, decreases in plasma and brain DHA levels have been shown in patients with AD. These results, however, remain controversial, since other studies have demonstrated an increase or an absence of variation in brain DHA levels in similar populations. Nonetheless, the risk of dementia was found to be augmented in elderly subjects presenting low levels of circulating EPA [184]. In addition, regular consumption of diets rich in n-3 PUFA, such as the Mediterranean diet, appears to contribute to a decrease in the risk of depression and/or dementia in the elderly [185, 186]. Using a mouse model of AD, the Tg2576 mouse, dietary supply of DHA reduces both the formation of amyloid plaques or the accumulation of caspase-cleaved products, while protecting against the loss of synaptic markers [187–189]. However, the administration of DHA-containing dietary supplements to patients with AD or mild cognitive impairment has not yielded conclusive results [190]. Only in vitro studies suggested a potential beneficial effect of DHA mediators. Indeed, it was shown in vitro that RvD1 promotes Aβ phagocytosis [191] and that NPD1 downregulates inflammatory signaling and amyloidogenic APP cleavage [192]. Data from preclinical and clinical studies all indicate that the effect of DHA differs according to apolipoprotein E (APOE) genotype [193–196]. A reduced brain uptake has been suggested as a mechanism underlying the lack of benefit of DHA in APOE4 carriers [193, 197]. However, APOE4 could simply counteract any effect of DHA by its known aggravating impact on peripheral immune and inflammatory responses, including microglial activation [198, 199]. Therefore, although preclinical studies support an effect of DHA in the prevention and / or treatment of age-related diseases, further clinical trials adapted to APOE genotype remain required to determine the best approach for an optimal therapeutic effect.
Growing evidence highlight an association between inflammation in cerebral tissues and mood and cognitive disorders during infection, aging and neurodegenerative disorders. Whether neuroinflammation processes are a cause or a consequence of the underlying disease and its symptoms is still debated. However, the current state of knowledge strongly points to potential disease modification value of treating neuroinflammation in the preclinical stages of neurodegenerative diseases. More specifically, the impact of DHA, either from the diet or from dietary-supplementation, as a preventive therapeutic strategy needs to be investigated.
Footnotes
Acknowledgments
This work has been supported by FRM, Région Aquitaine, Société française de Nutrition and INRA (SL). The work of FC is supported by grants from the Canadian Institutes of Health Research (CIHR), the Alzheimer Society Canada, the Parkinson Society Canada and the Fonds de la recherche en santé du Québec (FRQ-S)