
Abstract
We present a case of dichorionic-diamniotic twin females who developed hypoxemic respiratory failure. They were ultimately diagnosed by lung biopsy with alveolar capillary dysplasia with misalignment of pulmonary veins. This case highlights a practical approach to reaching a diagnosis in infants with suspected developmental lung disease.
Keywords
Introduction
Alveolar capillary dysplasia with misalignment of pulmonary veins (ACDMPV) is a rare fatal disorder caused by abnormal pulmonary vascular development [1]. This case report, to our knowledge, is the first in the literature of identical twins diagnosed with ACDMPV. We report their presentation and strategies in diagnosis and management.
Case report
Community hospital delivery room course
Dichorionic-diamniotic (DCDA) twin females were born at 37 weeks gestation to a 26-year-old G2P0 mother via cesarian-section following failure to progress. The prenatal course was uncomplicated with normal prenatal labs, imaging and without concern for infection at the time of delivery. Twin A was delivered limp and apneic, requiring three minutes of positive pressure ventilation (PPV) until she was transitioned to continuous positive airway pressure (CPAP) and weaned off respiratory support after 10 minutes of life. Apgar scores were 2, 6, and 8, and cord gas was notable for a metabolic acidosis (pH of 6.9 and base deficit of –11). By one hour of life, her capillary blood gas pH improved. Twin B (Birth Weight 2.55Kg) also had poor respiratory effort at birth, however, did not require PPV or other respiratory support, and her Apgar scores were 4 and 7. With improving respiratory assessments, the twins were transitioned to the well-baby nursery.
Twin A outside hospital course
The infant became hypoglycemic requiring glucose gel and formula supplementation while in the nursery. At 14 hours, she had increased work of breathing, with tachypnea, subcostal retractions and cyanosis. The baby was initially placed on CPAP with a fraction of inspired oxygen (FiO2) of 1.0 and subsequently intubated, given surfactant and transported to a Level III neonatal intensive care unit (NICU). There was no improvement in oxygen requirement after surfactant delivery.
On arrival to the NICU, she had an arterial blood gas with a pH of 6.79, a pCO2 of 77, paO2 of 62 with a lactate of 12 and a base deficit of –23. Initial chest x-ray showed bilateral patchy hazy lung fields. (Fig. 1) Given persistent need for FiO2 of 1.0, severe respiratory and metabolic acidosis and hypotension, high frequency jet ventilation, inhaled nitric oxide (iNO) at 40 ppm and epinephrine infusion were started, and multiple crystalloid boluses were administered. An echocardiogram (ECHO) showed normal segmental anatomy with evidence of pulmonary hypertension (PH), including right to left shunting at both the ductal and atrial levels. Given persistent PH as noted by 10% pre- and post-ductal differential saturations, sedation and muscle relaxation were also used. Due to persistent hypoxemia at three days of life, she was transferred to a Level IV NICU with extracorporeal membrane oxygenation (ECMO) capability.
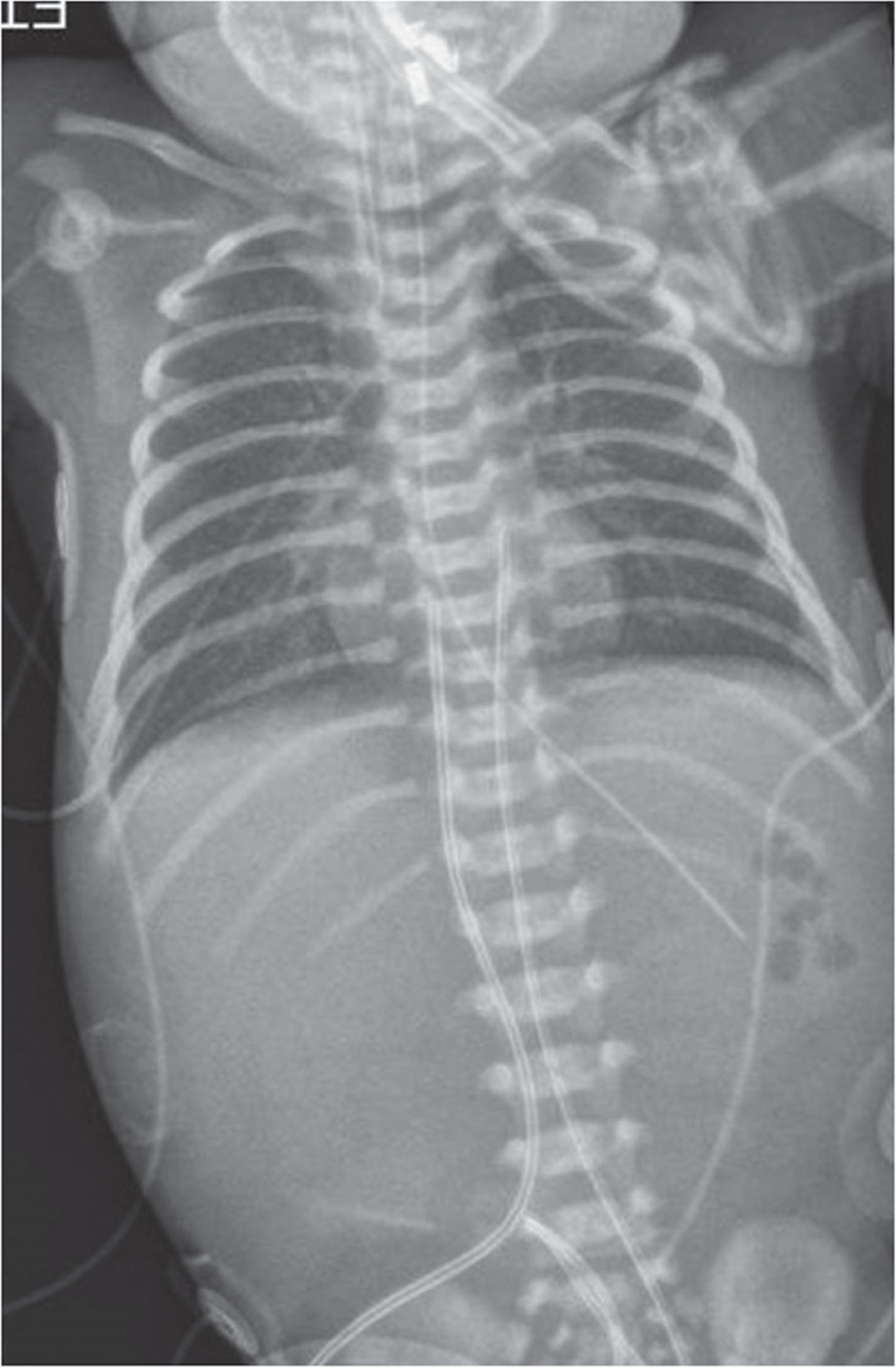
Chest X-Ray of Twin A: Mild hyperinflation and nonspecific hazy opacity of both lungs. No focal consolidation, pleural effusion or pneumothorax.
At about 16 hours, she became cyanotic with increased work of breathing, requiring PPV, intubation and transfer to a Level III NICU. Initial support included minimal conventional ventilatory setting with FiO2 requirements around 0.4. At four days of life, oxygen requirement increased and iNO was initiated at 40 ppm. An ECHO showed normal segmental anatomy, decreased right ventricular (RV) function, suprasystemic RV pressures evidenced by severe posteriorly bowed ventricular septum and right to left shunting at the atrial level. There was no patent Ductus Arteriosus (DA) flow. Epinephrine, milrinone and epoprosterol were initiated. She was transferred to a Level IV NICU at five days for further management.
Level IV NICU course
Twin A was cannulated to veno-arterial (VA)-ECMO within two hours of arrival to the ECMO center due to persistent hypoxemia. She had an arterial blood gas with a pH of 7.34, a pCO2 of 35, paO2 of <34 with a lactate of 2.4 and a base deficit of –6. Rapid genetic labs, which require blood samples from both parents and the infant were sent, including whole exome sequencing (WES) panel for developmental disorders of the lung and PH. She was successfully decannulated after four days of ECMO support at age seven days, however her vessels were not suitable for reconstruction, precluding the possibility of repeat cannulation. Upon decannulation, conventional ventilation settings were optimized, she remained on iNO of 20 ppm with milrinone at 0.5mcg/kg/min and epinephrine at 0.03 mcg/kg/min. Sildenafil was initiated and up-titrated to 1 mg per Kg per dose every eight hours, with each dose of sildenafil, iNO was weaned and ultimately weaned to 5 ppm. A Chest CT obtained at 12 days had non-specific increased lung parenchymal findings, with a few small cysts and thickened septa in the upper lobes.
Twin B was managed on conventional ventilation and iNO, with FiO2 of 0.4–0.6. An ECHO displayed severe PH, with all right to left shunting at the foramen ovale and no PDA. Prostaglandin E was initiated, and ductal patency was achieved successfully; inhaled iloprost was started to further decrease the pulmonary vascular resistance but subsequently weaned with the introduction of sildenafil. By 11 days, she was weaned off iloprost, reached goal dose sildenafil and completed a course of antibiotics for culture negative sepsis. On day 12 she had an acute decompensation in the setting of multiple pulmonary hypertension crises that briefly responded to sedation and reinitiating iloprost. However ultimately, she was canulated onto VA ECMO after a sustained desaturation that was refractory to sedation, escalation in ventilatory support and iloprost boluses.
By day13, Twin A had been decannulated and was experiencing a period of relative clinical stability on nasoduodenal feeds, enteral sildenafil and 5PPM of iNO. Twin B was on day two of VA-ECMO. Rapid WES of both twins was negative and prompted an open lung biopsy to obtain a diagnosis. Which twin to biopsy was discussed at length, as the procedure could cause a significant clinical decompensation. Twin A was unable to be re-cannulated back onto ECMO due to vessel ligation after her first ECMO run. Twin B, on ECMO, would be supported during surgery, though was significantly anticoagulated with increased risk of procedural bleeding. The risk of bleeding for Twin B outweighed that of clinical decompensation of Twin A, and Twin A underwent an open lung biopsy. In preparation, iNO was increased to 20 ppm and bolus dose of iloprost was available during procedure should they have a PH crisis. Twin B was decannulated on ECMO day three and remained on iNO of 20 ppm.
The lung biopsy was obtained without significant clinical decompensation. ACDMPV diagnosis was rendered by the institutional pathologist and confirmed by a pediatric pathologist with expertise in developmental lung disease (Fig. 2).
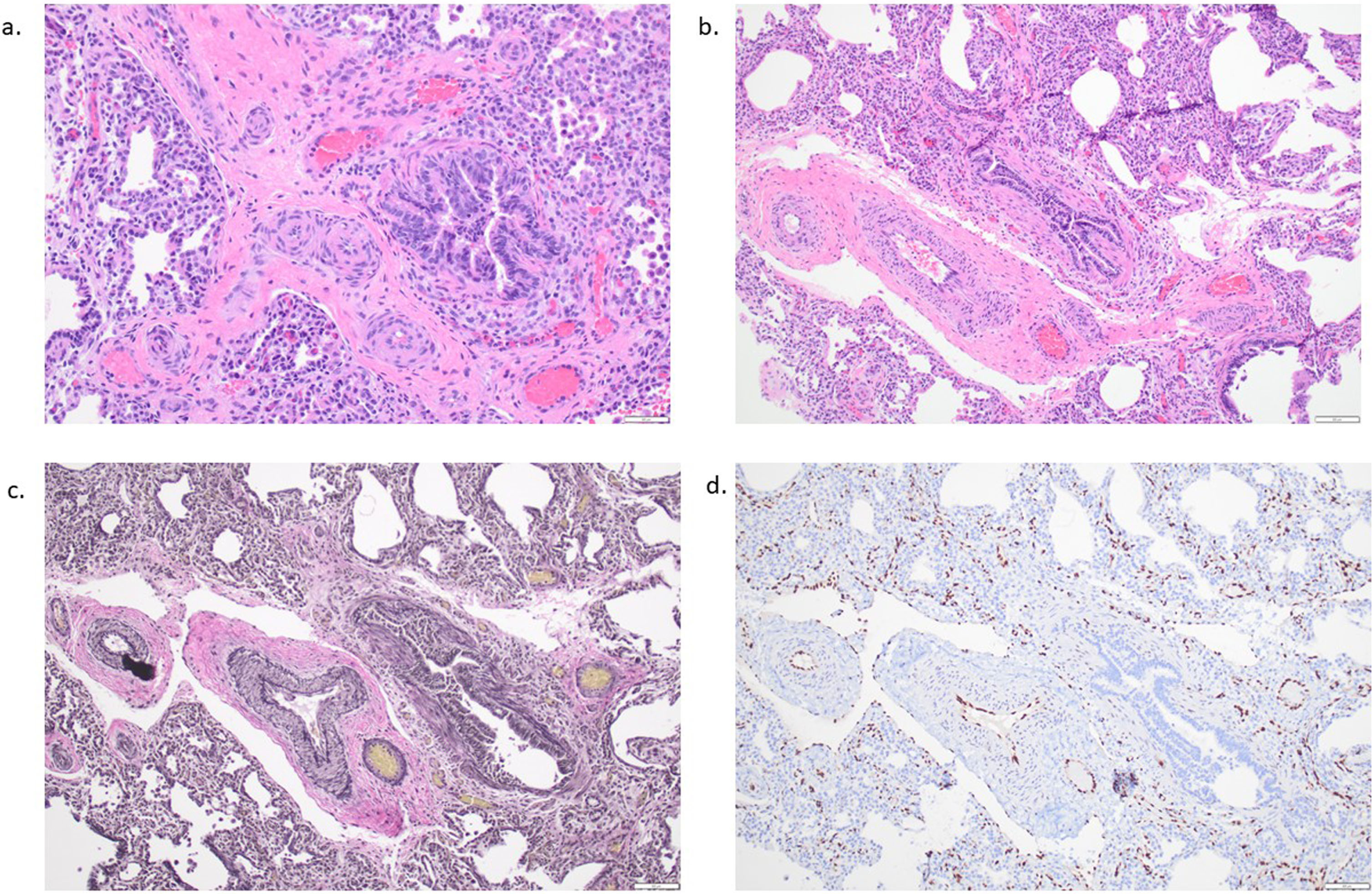
A) Alveolar lung parenchyma with diffusely thickened alveolar walls (hematoxylin and eosin stain, 20x), B) pulmonary arteries/arterioles with medial hypertrophy adjacent to misaligned pulmonary veins (hematoxylin and eosin stain, 100x) C) highlighted with elastic stain (elastic stain, 100x) and D) capillaries demonstrating absence of contact with overlying epithelium (ERG immunohistochemical stain, 100x).
Following the diagnosis, lung transplant candidacy was discussed with the family. Neither was considered a candidate by the neonatal lung transplant center, due to age, size, likelihood of disease progression and vessel ligation following decannulation from ECMO. Furthermore, the severity of their disease decreased the likelihood of survival until transplant.
A neonatal palliative care consult was obtained. The parents opted to continue mechanical ventilation and iNO but opted to make the twins DNR should they go into cardiac arrest. Twin B developed persistent hypoxemia, despite iNO and iloprost on day of life 24. At this time, the parents redirected care and the breathing tube was removed. Twin A remained on minimal conventional vent support and iNO at 5 ppm. On day of life 33, she had increasing oxygen requirements and persistent hypoxemia, resulting in her passing on day of life 35.
Though the twins were DCDA, the genetics team was able to confirm that they were identical, allowing the team to extrapolate Twin A’s biopsy results to Twin B. Further genetic analysis did not uncover any underlying genetic mutation.
ACDMPV is a rare fatal disorder caused by abnormal pulmonary vascular development, which causes significant diffusion defects. Due to the patchiness of ACD within the lungs, patients may present at various times during the neonatal period or even much later [1]. Disease progression can also be variable with initial periods of improvement and responsiveness to pulmonary vasodilator therapy with ultimate refractory hypoxemia and death. This case, to our knowledge, is the first in the literature of identical twins diagnosed with ACDMPV.
Over the past several years our center has had the opportunity to take care of multiple patients with developmental lung disease. Based on our experience, we have developed a workflow to reach a diagnosis when potential lethal developmental lung disease of the newborn is suspected (Fig. 3). The first step is rapid WES for all patients. While genetic results are pending, a chest CT is obtained to help further elucidate underlying pathology and allow for potential surgical planning to identify optimal biopsy target if genetic studies are inconclusive. Because of the known biopsy risks associated with open lung biopsy in these vulnerable infants, minimally invasive procedures are prioritized.
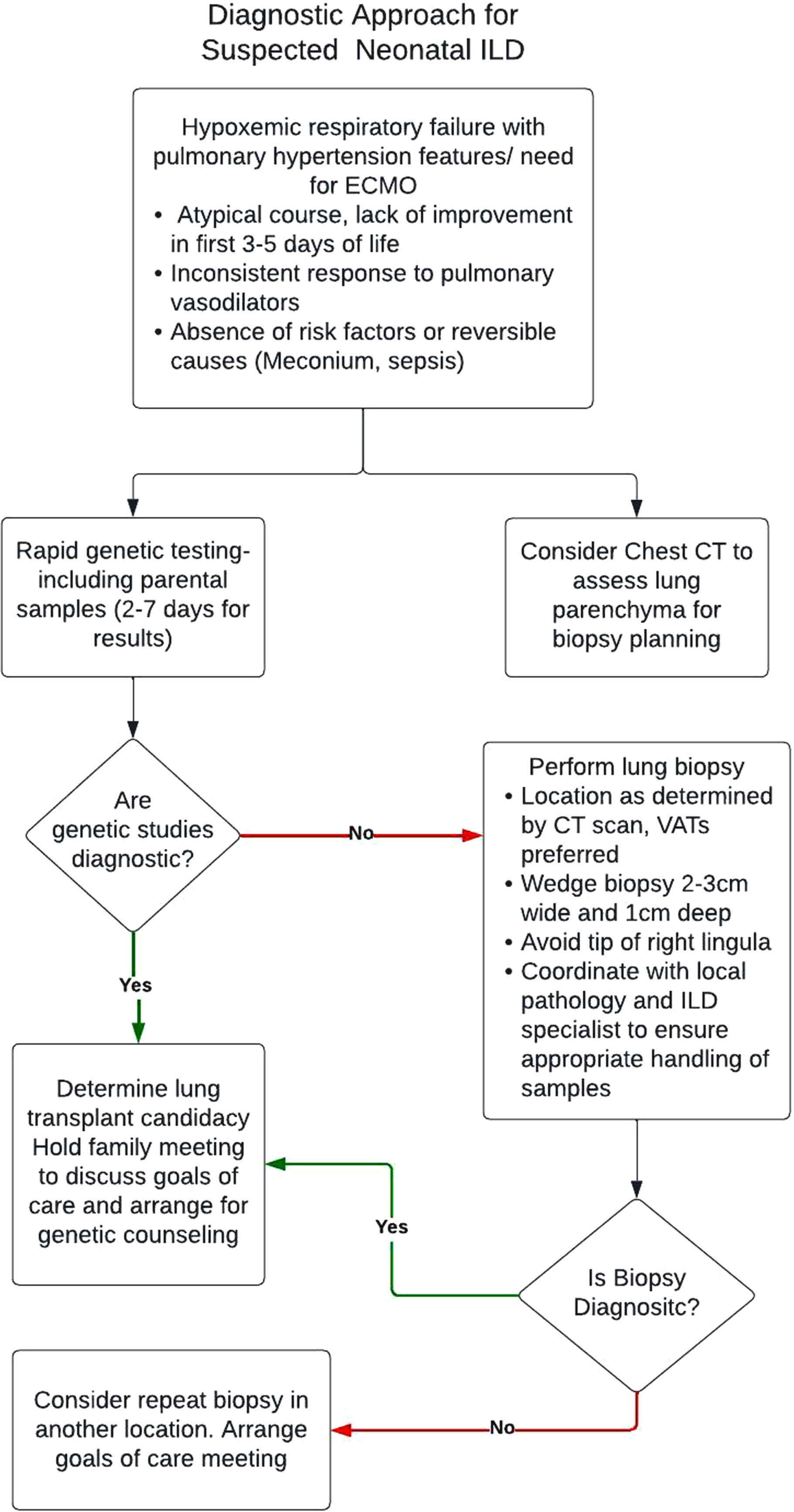
Diagnostic approach for suspected neonatal ILD.
The delivery room presentation in this case is interesting as the twins required some resuscitation at birth, with Twin A having a more severe presentation with both an acidotic cord gas and need for several minutes of PPV. This is not common in patients with ACDMPV, as 95% of these patients will have normal birth weights and Apgar scores [2]. Furthermore, Twin A experienced hypoglycemia, which is not usually associated with ACDMPV. Together these findings lead to a strong initial suspicion for early onset sepsis as the cause of the twin’s respiratory failure. ACDMPV should be considered if persistent pulmonary hypertension (PPHN) does not improve within the first two weeks. Our team took over the care of the twins at three and five days of life. Given their lack of clinical improvement despite antibiotic therapy, the bilateral hazy lung fields on chest x-ray and a period of clinical stability in the well-baby nursery prior to clinical decompensation, a genetic workup for developmental lung disease was initiated.
As described previously in the literature, each twin had an underlying reactive pulmonary vascular bed responsive both to iNO and prostacyclin therapy [3]. Unlike other cases of ACDMPV, neither twin developed pulmonary edema with iNO therapy or oral sildenafil therapy [4]. Though Twin A was initially the sicker of the two, Twin B ultimately passed prior to her sister, after developing progressively worse PH unresponsive to prostacyclin or iNO. Of note, Twin B had a small, constricted DA despite prostaglandin therapy, whereas Twin A had an open DA throughout her course, which potentially allowed her to better recover from PH crisis events as compared to her sister.
There have been over 200 cases of ACDMPV documented within the literature and approximately 60% have had variants in the forkhead box F1(FOXF1) gene [5]. The mode of transmission is mainly de novo [6]. FOXF1 is thought to be partially paternally imprinted as many of the mutations have occurred on the maternal allele [5]. Extrapulmonary malformations, most commonly cardiac defects, are reported in 50% –80% of cases, most of which have an identified mutation in the FOXF1 gene [7]. Though a normal genetic result does not exclude the diagnosis of ACDMPV, it is an important first step that can obviate a diagnostic open lung biopsy. Furthermore, if a gene is identified it can be helpful for family planning as it can identify a carrier parent. Even in the case of de novo mutation some recommend prenatal testing for future pregnancies as there is a risk of recurrence due to germinal mosaicism in one parent [6]. Though genetics were normal in this case, genetic analysis was important as it confirmed that the DCDA twins were identical. Monozygotic twin births represent approximately a third of all twin deliveries and of those that are monozygotic twins, 25% will be DCDA twins [8].
Without a genetic diagnosis, histopathologic diagnosis is required. The most common histopathologic features of infants with ACDMPV include reduced and dysplastic alveoli, pulmonary artery and vein within the same bronchovascular bundle, and thickened alveolar septa with deficiency of alveolar capillaries [9]. Twin A underwent a left upper lobe wedge biopsy, which showed alveolar lung parenchyma with abnormal lobular architecture, including thickened alveolar walls. The pulmonary arteries/arterioles with medial thickening were adjacent to misaligned pulmonary veins within the same adventitial sheet. Additionally, the capillaries were abnormally located within the central portion of the alveolar walls rather than adjacent to epithelial cells.
Though lung biopsy is considered the gold standard for diagnosis of ACDMPV, a falsely negative biopsy can occur due to sampling bias which is more likely to occur in patients with patchy lung involvement and more atypical presentations [10]. To improve diagnostic accuracy and aid in surgical planning all patients at our center undergo a CT chest prior to the biopsy to help target an area that will most likely provide tissue that will aid in diagnosis. That said, the twins presented in this case likely had diffuse lung involvement given their severe early onset presentation, thus the chances of obtaining a falsely negative biopsy were low.
Classical ACDMPV which is diagnosed within the first month is almost always a lethal diagnosis, with most patients dying within the first month of life [11]. However, patients with atypical presentations who present later in life or with less aggressive disease, can survive till lung transplantation. In one case series published by a neonatal lung transplant group, six patients with atypical ACDMPV, underwent lung transplant, with similar one and five year survival rates as compared to infants transplanted for other indications [12]. The twins in this case were not considered to be transplant candidates because of the severity of their disease and the unlikelihood that they would survive the expected four to six month waiting period for a lung transplant.
This is the first case of twins with ACDMPV that has been reported in the literature. A multidisciplinary approach utilizing early clinical suspicion, rapid genetics and lung biopsy when genetics are inconclusive is crucial in obtaining a diagnosis for these patients and helps with effective counseling of families.
Disclosure statement
The authors declare no conflicts of interest.