
Abstract
The incidence and prevalence of chronic kidney disease (CKD) and end stage renal disease (ESRD) are on the rise causing significant health issues associated with hemodialysis, peritoneal dialysis and kidney transplantation. Moreover, all CKD patients have dramatically higher risk than the general population of developing kidney tumors with 5% incidence of renal cell carcinoma. This review highlights the spectrum of precursor lesions, most common benign and malignant neoplasia in patients with CKD and ESRD, focusing on pathogenesis, morphologic and immunophenotypic findings.
INTRODUCTION
Chronic kidney disease (CKD) and end stage renal disease (ESRD) represent a public health issue with severe consequences for the patient, including diminished quality of life and life expectancy [1]. In developed countries, including the United States, CKD and ESRD are mainly secondary to diabetes and hypertension with associated cardiovascular risk factors [1, 2]. Other causes, like glomerulopathies, are more common in Asia and other regions of the world [3]. The Kidney Disease Improving Global Outcomes (KDIGO) Foundation guidelines define chronic kidney disease (CKD) stages based on biomarkers of kidney damage and proteinuria. ESRD, represents the final stage of CKD, and is defined as a glomerular filtration rate (GFR) of less than 15 mL/min [4, 5], with subsequent need for renal replacement therapy (RRT) or kidney transplant.
Despite improvements in detection of CKD, the incidence and prevalence of ESRD is estimated to further increase by the end of the current decade [6]. The standard of care for the treatment of ESRD is renal transplantation, however, most patients with ESRD will undergo hemodialysis or peritoneal dialysis and remain on these RRT modalities while awaiting to receive a kidney transplant. In the United States, the waiting times for receiving a kidney transplant are variable. For example, in 2021, almost half of the patients had to wait 2 or more years to receive a kidney transplant [7]; of these, approximately 10% remained in the waiting list for more than 5 years. This long waiting time is due to an increasing demand and concurrent donor organ shortage [8]. In regions of the world without a robust system of organ donation, most patients that are able to access RRT will remain in dialysis without receiving a transplant [3].
In general, all patients with CKD have an increased risk of developing RCC, although the risk increases significantly with the development of ESRD and more strongly with the time spent receiving dialysis treatment [9, 10]. It has been estimated that the likelihood of developing renal cell carcinoma is up to 100 higher than in the general population [11–13]. This increased risk translates to an incidence of renal cell carcinoma of approximately 3–7% [14]. The patients who have received kidney transplantation also have increased risk of developing malignancies, including RCC, in comparison to the general population [15].
This review is focused on two most frequent tumors in CKD/ESRD patients, namely acquired cystic disease-associated renal cell carcinoma (ACD-RCC) and clear cell papillary renal cell tumor (CCPRCT), as well as common precursor lesions.
ACQUIRED CYSTIC KIDNEY DISEASE-ASSOCIATED RENAL CELL CARCINOMA (ACD-RCC)
Renal cell carcinoma in the setting of ESRD can be present with or without acquired cystic kidney disease. Acquired cystic disease (ACD) has been defined as the presence of 3 or more cysts per kidney, or cysts accounting for > 25% of the renal parenchyma in a patient without polycystic kidney disease [16, 17]. The development of cysts becomes more obvious and extensive in cases of ESRD, but ACD can be present in CKD before the development of ESRD and need for dialysis [18–20]. Transplantation reduces the severity of renal parenchyma involvement by ACD [21]. It has been proposed that the cyst formation process is due to progressive destruction of kidney parenchyma by uremia, oxidative stress, mitochondrial DNA mutations and oxalate accumulation inducing hyperplasia of the residual nephrons, becoming a trigger of the degenerative process related to subsequent cyst formation [20, 22].
There are some limitations to the definition of ACD. The studies that have established the criteria are initially based on imaging, but there are also studies that had utilized pathologic findings from nephrectomies, and kidneys obtained from autopsies. However, none of these studies established a required minimum of cyst size. The 3 or more cyst criteria is also not fully applicable in the pediatric populations, as they may present with a lesser of number of cysts than in the adults [19].
In clinical practice, the diagnosis is performed by utilizing imaging studies, either by ultrasound (US), computed tomography (CT), or magnetic resonance imaging (MRI). The current guidelines of the American College of Radiology (2020) are supportive of use of ultrasound as the most appropriate initial imaging for patients with kidney failure, either acute or chronic [23]. CT and MRI require contrast agents with a more careful evaluation of the risk-benefit relationship in the context of renal failure [24], and generally reserved for follow up and detection of complications [20]. There are no clear guidelines regarding patient screening to detect ACD and associated malignancy, other than performing imaging screening to potential transplant candidates [25].
During follow-up, the evaluation of cystic masses for the purpose of prediction of cancer likelihood and decision making relies on the utilization of the Bosniak Classification primarily based on CT or MRI scans, but not US [26]. During the surveillance of ACD, most tumors are detected incidentally. Most ACD-RCC are initially managed with surveillance or non-radical approached, as most cysts will fall within Bosniak IIF and III categories [27].
ACD-RCC was recognized as an independent entity since the 2016 edition of the WHO Classification of Tumors Urinary and Male Genital Tumours [28]. Before WHO classification update, many of these tumors were diagnosed as RCC-unclassified or as ‘type 2’ papillary RCC (PRCC) [12], the latter is a diagnosis currently not utilized according to the latest 2022 edition of the WHO [29].
Per definition, the ACD-RCC is present in a background of ACD. The duration of dialysis is related to an increased risk and incidence of ACD-RCC, with an overall incidence of 50%, and increasing cumulative incidence of 13%, 50% and 85% at 2, 6, and 9 years, respectively [20, 22]. Another review estimated ACD-RCC incidence as 24 times higher when compared to the general population without ESRD [30].
Macroscopically in ACD patients kidneys display multiple unilocular and multilocular cysts containing clear, straw-colored, or gelatinous fluid, with frequent hemorrhage [13, 22, 31, 32]. The ACD-RCC tumor may be present as a combined solid/cystic mass, including some that are clearly arising from a cyst, or pure solid masses with an average size of 4 cm [33]. The presence of solid growth is required for a diagnosis [31]. There is frequent multifocality and bilaterality, in 50% and 10–12% of cases, respectively [13, 33, 34]. In addition, concurrent neoplasms may coexist with the tumor [31].
Microscopically ACD-RCC are quite heterogeneous tumors composed of eosinophilic or clear cells with intracytoplasmic vacuoles, high-grade nuclei (majority grade 3), and organized in various architectural patterns including tubular, papillary, microcystic, cribriform and solid (Fig. 1). However, the most characteristic features are presence of cribriform or “sieve-like” architecture, and an abundant deposition of intratumoral calcium oxalate crystals [22, 31] which are seldom found in other tumor types [14, 35]. ACD-RCC may rarely present with rhabdoid or sarcomatoid differentiation, features associated to a worse prognosis in a tumor that usually has rather indolent clinical course [16, 30, 33]. Immunohistochemical studies may aid in the diagnosis, albeit they are mostly useful to confirm the tubular proximal cell origin due to diffuse expression of PAX8, AMACR and CD10, and negativity for CK7, CKIT and GATA3 [16, 22, 33].

Acquired cystic disease associated renal cell carcinoma (ACD-RCC). Gross photograph of dramatic example of ACD with extensive cystic change and hemorrhagic mass at the bottom right (A). Low magnification image showing heterogeneous architectures combining papillary, cystic, tubular and solids patterns (B). Sieve-like or cribriform morphology is the most characteristic growth pattern (C). Predominantly eosinophilic cells with prominent nucleoli have intracytoplasmic or intercytoplasmic lumina and holes imparting sieve-like architecture (D). Immunohistochemistry in ACD-RCC is typically positive for P504 (E), CD10 and RCC (not shown), whereas negative for CK7 (F), CKIT and GATA3 (not shown). Intratumoral oxalate crystals could be abundant like in these two cases (G-H), but are not required for the diagnosis.
ACD-RCC cases have recurrent, often coexisting, genomic alterations in KMT2 C and TSC2 genes [36]. Some of historic ACD-RCC cases may in fact represent TSC-mutant renal cell tumors like ESC-RCC developing in patients with tuberous sclerosis complex and CKD/ESRD [37]. Chromosomal structural abnormalities have also been described in ACD-RCC, including gain of chromosomes 3, 7, 16, and X, and loss of chromosome Y [34], although these findings are not specific and can be seen in other RCC subtypes and may be also present in non-neoplastic kidney tissue in ESRD cases [38].
RENAL CELL TUMORS ARISING IN CKD WITHOUT ACQUIRED CYSTIC KIDNEY DISEASE
All other tumors arising in the context of CKD without ACD, show morphologic and genomic similarities to the tumors arising sporadically in kidneys of patients without CKD. Therefore, and except for the discussion of clear cell papillary renal cell tumor (CCPRCT), the morphologic features used for the diagnosis of these tumors will not be discussed on this article, as only their incidence and distribution vary among these groups of patients.
The entity that has been traditionally associated to the presence of ESRD is CCPRCT. In a 2006 study by Tickoo et al [31], in addition to describing ACD-RCC, the presence of CCPRCT was also described in cases of ESRD, involving up to 21% of the cases of their study. Initially, it was considered a tumor with higher prevalence in ESRD, however, it was later demonstrated that CCPRCT incidence in kidneys with ESRD may be similar or even lower to the kidneys without ESRD [39, 40]. CCPRCT is generally detected incidentally and has overwhelmingly favorable outcome with exceedingly rare reports of metastases or high stage features [41, 42]. To emphasize indolent behavior of CCPRCT, their designation was changed from “carcinoma” to “tumor” in the most recent 2022 WHO classification [29].
CCPRCT are typically encapsulated and organ confined small tumors averaging 2.5 cm with variable solid or cystic components, but could be multifocal and bilateral in 10–25% cases [39, 43, 44]. Microscopically, CCPRCT have heterogeneous architecture with cystic, papillary and acinar areas, as well as prominent branching anastomosing tubules, thus an alternative nomenclature of “clear cell tubulopapillary” renal cell tumor was proposed as more appropriate and distinctive from other clear cell and papillary tumors [43]. The tumor cells have clear cytoplasm and low grade nuclei (grades 1–2) which tend to be aligned away from the basement membrane in a linear fashion [22, 39, 45] (Fig. 2). CCPRCT origin from distal nephron is highlighted by HMWCK and/or GATA3 co-expression in addition to characteristic diffuse co-expression of CK7 and CAIX (cup-like) [43, 46, 47]. Low stage and low grade typical morphology coupled with lack of chromosomal abnormalities/VHL mutations considered essential for CCPRCT diagnosis.

Clear cell (tubulo)papillary renal cell tumor (CCPRCT). Small tumor with thick capsule (A); clear cell morphology with low grade nuclei aligned in linear fashion (B). Co-expression of CAIX (C) and CK7 (D) is a hallmark immunoprofile of CCPRCT. Expression of HMWCK (E) and GATA3 (F) is present in the vast majority of cases indicating CCPRCT origin from distal nephron. Anastomosing and branching tubules with (pseudo)papillary architecture is a very common finding (G) supporting tumor renaming to clear cell tubulopapillary renal cell tumor. Typical cup-shaped CAIX expression due to lack of staining at the apical aspects of tumor cells (H).
The incidence of other carcinomas in patients with CKD/ESRD without a background of ACD, follows this order in descending frequency: clear cell renal cell carcinoma (CCRCC), PRCC, and chromophobe RCC. In order of frequency, although not a carcinoma, CCPRCT would be the fourth most common entity [39, 44].
BENIGN TUMORS AND PRECURSOR LESIONS ARISING IN KIDNEYS OF CKD PATIENTS
Multiple risk factors are present for the development of kidney neoplasia, including an abnormal tissue environment with increased oxidative stress, upregulation of growth factors and other cytokines [22]. Furthermore, in case of cyst formation, the epithelial changes with proliferation, atypia and fluid cyst contents rich in catabolites and cytokines are also contributors to the neoplastic process [20, 48]. These factors create a proinflammatory and profibrotic microenvironment that induces neoplastic changes [49].
There are several questions that remain open regarding neoplastic precursor lesions in the kidney. Some precursor lesions have been well studied, such as the papillary adenoma-carcinoma sequence, the development of cysts and progression towards carcinoma. Other tumors remain without a suspected or determined precursor, or as in the case of CCPRCT, there is no size criteria to distinguish a neoplastic precursor versus a fully developed tumor [39].
The most common tumor found in all cases, with or without ESRD, is papillary adenoma. These tumors must measure equal or less than 15 mm in diameter to be considered adenomas. This neoplasm has been accepted as PRCC precursor, as it shares molecular and cytogenetic alterations with PRCC [50, 51].
Other known precursor lesion is the development of retention cysts due to progressive destruction of kidney parenchyma with alternating atrophy, fibrosis and hypertrophy of the residual nephrons [20]. Some of these cysts develop atypical epithelial features, including complex architecture with papillae formation, epithelial stratification or eosinophilic vacuolated cell lining with multilocular ACD-RCC-like appearance [52–54]. The architectural patterns, crystal depositions and cytologic features of cyst lining have been linked to CCPRCT, ACD-RCC, and CCRCC [46, 52–54], or could be characterized as recently described papillary renal neoplasm with reverse polarity (Fig. 3). Another potential precursor lesion of “distal tubular hyperplasia” forming hyperplastic tufts or small inflammatory polyps has been recently described, however no neoplastic counterpart has been established yet in association with this lesion [55].
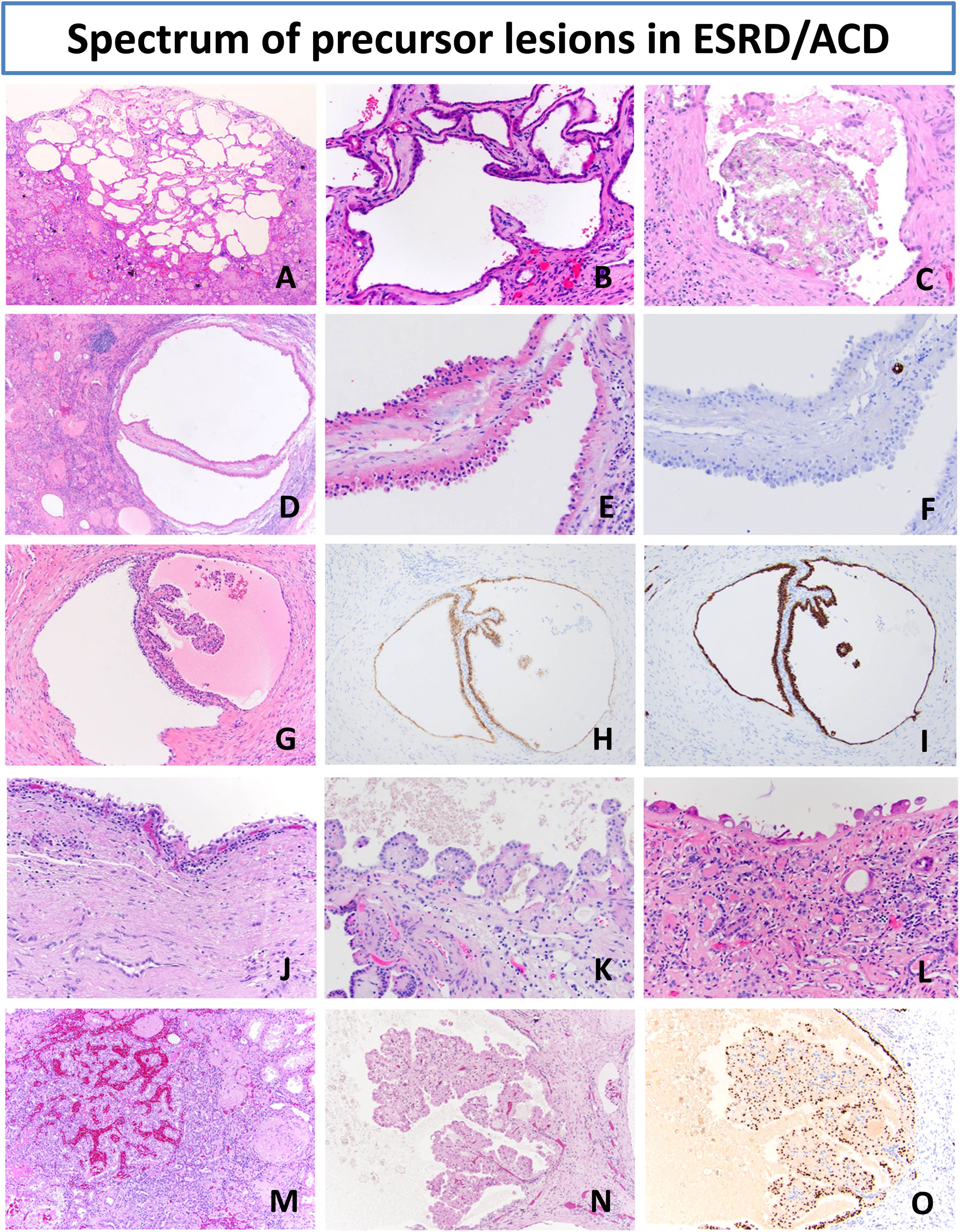
Precursor lesions in end-stage renal disease (ESRD) and acquired cystic kidney disease (ACD). The most common finding in ACD is presence of variably sized unilocular cysts and small clusters of multilocular cysts (A-B) often with oxalate crystal deposits (C). Background kidney of ACD-RCC patient with atypical renal cyst with eosinophilic stratified lining negative for CK7 (D-F). Background kidney of ESRD patient with CCPRCT precursor cysts (G) with clear cell lining, small papillary projects and co-expression of CAIX (H) and CK7 (I). ESRD kidneys with different types of cysts: clear cell type (J), atypical eosinophilic papillary type (K) and atypical eosinophilic non-stratified hobnailed lining (L). Microscopic papillary adenomas (M) are the most common benign tumors in ESRD setting, however recently described entity of papillary neoplasm with reverse polarity could also arise in ESRD and is characterized by delicate papillary projections of bright eosinophilic cells with low-grade polarized nuclei (N) and characteristic GATA3 expression (O).
CONCLUSION
The main cause of death in patients with CKD and ESRD are cardiovascular complications, however, it is important to recognize the increased risk for the development of renal cell neoplasms, including some with malignant potential, which is an important factor added to the morbidity and potentially affecting quality of life and survival of patients with CKD and ESRD.
Footnotes
ACKNOWLEDGMENTS
The authors have no acknowledgments.
FUNDING
The authors report no funding.
AUTHOR CONTRIBUTIONS
Both authors contributed to study design, conception of idea, performance and interpretation of reviewed literature; as well as manuscript writing.
CONFLICT OF INTEREST
David Henriquez Ticas and Maria Tretiakova have no conflicts of interest to report.