
Abstract
SMARCB1-deficient renal medullary carcinoma is one of the most aggressive forms of kidney cancer and mainly affects individuals with sickle hemoglobinopathies. Herein, we have sought to provide a contemporary update regarding our understanding of this entity. Recent studies have improved our understanding of the pathophysiology of this tumor including potential modifiable risk factors, putative cell of origin, and mechanisms of SMARCB1 loss. Furthermore, mechanisms of oncogenesis based on the SMARCB1-deficient phenotype continue to be elucidated. These tumors are classified as SMARCB1-deficient renal medullary carcinoma, and SMARCB1-deficient medullary-like renal cell carcinoma based on the presence of sickle hemoglobinopathies, and established subtypes of renal cell carcinoma with secondary SMARCB1 loss continue to be identified. Current diagnostic algorithms help in separating the above-mentioned tumors from morphologic mimics which include collecting duct carcinoma, upper tract urothelial carcinoma, FH-deficient, and ALK-rearranged renal cell carcinoma which have different natural histories and therapeutic implications. Finally, there is an unmet need to develop better treatment regimens for patients with renal medullary carcinoma given its aggressive clinical course and several agents continue to be evaluated in addition to standard-of-care platinum-based cytotoxic chemotherapy.
Keywords
INTRODUCTION
SMARCB1-deficient renal medullary carcinoma is one of the most aggressive forms of kidney cancer and predominantly affects individuals with sickle hemoglobinopathies. Significant strides have been made in the past decade towards understanding the pathophysiology of this tumor, refining diagnostic criteria, and several clinical trials are underway to develop better therapeutics. Herein, we have sought to provide a contemporary update regarding our understanding of this neoplasm.
EPIDEMIOLOGY
Renal medullary carcinoma is a high-grade adenocarcinoma with SMARCB1 deficiency and comprises less than 0.5% of all renal cell carcinomas [1–3]. It typically occurs in patients with sickle hemoglobinopathies, with the most common being sickle cell trait [2, 4].
However, the prevalence of renal medullary carcinoma amongst patients with sickle cell trait is not particularly high, with an estimated prevalence of 5 in 100,000 [5]. However, under-reporting may obscure accurate estimates of renal medullary carcinoma incidence, particularly in areas such as Africa, where the immunohistochemistry needed to establish SMARCB1 loss may not be available [6]. For example, in a survey of patients with sickle cell disease from Nigeria, where sickle cell trait is estimated to be present in excess of 20% of all births [7], only 0.056% (2 of 3596) patients were diagnosed with renal medullary carcinoma over a 10-year period and none of these cases were confirmed by SMARCB1 immunostaining [8]. In the absence of accurate renal medullary carcinoma prevalence estimates in resource-limited settings, the possibility that environmental or other locoregional factors contribute to increasing renal medullary carcinoma incidence among individuals with sickle hemoglobinopathies in the United States and Europe cannot be excluded. Notably, amongst patients with sickle cell trait and renal medullary carcinoma there is a lack of bilaterality, multifocality, and familial clustering [1, 9, 10]. It has therefore been proposed that “environmental rather than genetic factors” may serve as important risk factors in the development of renal medullary carcinoma in patients with sickle cell trait [10].
CLINICOPATHOLOGIC FEATURES
Patients diagnosed with renal medullary carcinoma have a median age at presentation of 21–35 years, with a strong predilection for male patients with sickle hemoglobinopathies [2, 4, 11–17]. The most common presenting symptoms are hematuria and flank/abdominal pain, followed by abdominal mass, weight loss, and metastatic disease [1, 2, 11, 13].
In 1995 Davis et al proposed renal medullary carcinoma as the seventh sickle cell nephropathy [11], after the original description of nephropathies in patients with sickle cell trait by Berman et al in 1974, which included hematuria, papillary necrosis, nephrotic syndrome, renal infarction, the inability to concentrate urine, and pyelonephritis [3, 18]. The majority of patients with renal medullary carcinoma carry the (heterozygous) sickle cell trait [hemoglobin (Hb) AS], while in rare instances these tumors have been reported in patients with compound heterozygous sickle hemoglobinopathies (HbSC disease, HbS/ß-thalassemia), as well as in patients with (homozygous) sickle cell disease (HbSS) [5, 11, 13, 19]. This may be due to the much higher prevalence of sickle cell trait in the population compared with all other sickle hemoglobinopathies. If we assume that all sickle hemoglobinopathies confer the same renal medullary carcinoma risk, then we would expect that for every 55 patients with renal medullary carcinoma carrying the sickle cell trait, there would be only one patient with sickle cell disease [17].
PATHOPHYSIOLOGY
It has been estimated that the hypoxemic microenvironment within the renal medulla (partial pressure of oxygen of less than 40 mm Hg) is below the threshold at which HbS polymerizes (45 mm Hg) and promotes sickling of red blood cells in all sickle hemoglobinopathies, including sickle cell trait [3, 17, 20]. The consequent chronic hypoxemia is central to the development of sickle cell nephropathy.
Furthermore, renal medullary carcinoma is perhaps one of the few primary renal tumors with a marked predilection for the right kidney. Even in the initial description of renal medullary carcinoma by Davis et al, 23 of 31 (74%) of the reported tumors were on the right side [11]. Similarly, more recent cohorts diagnosed using contemporary criteria have reported right sided renal medullary carcinoma in 70 to 76% of cases [4, 10, 21]. This has been attributed to the increased length of the right renal artery and its contributory role in further promoting ischemia in the renal medulla [10, 17].
In addition, a recent case-control study has proposed that high-intensity exercise in patients with sickle cell trait may represent an additional (modifiable) risk factor for the development of renal medullary carcinoma [10]. Patients with renal medullary carcinoma had significantly higher rates of “positive activity index, reported exercise, athletic involvement, and military service” (correlated with higher skeletal muscle surface area) compared to matched controls [10]. These findings were further supported by genetically modified mouse models carrying the sickle cell trait that exhibited significantly higher hypoxia, particularly in the right kidney, following high-intensity exercise compared with wild-type control mice. Conversely, moderate intensity reduced renal hypoxia compared to the resting phase in both sickle cell trait and control mice [10]. Although these findings require further validation, they are consistent with safe exercise recommendations for patients with sickle cell trait that have been generated by multiple professional bodies and should be considered for patient counselling [10].
At the molecular level, recent studies suggest that in a physiologic state hypoxia-induced degradation of SMARCB1 helps adapt to the hypoxemic microenvironment of the renal medulla [22]. Furthermore, tumor cells that develop a SMARCB1-deficient phenotype secondary to prolonged exposure to such a microenvironment develop a distinct survival advantage that promotes oncogenesis [22].
The SMARCB1 gene (also known as INI1, SNF5, and BAF47) on the chromosome 22q11.23 locus encodes for one of the components of the evolutionarily conserved SWI/SNF (SWItch/Sucrose Non-Fermentable) chromatin remodeling complex [23]. The current edition of the World Health Organization (WHO) classification of renal tumors classifies SMARCB1-deficient renal medullary carcinoma as a molecularly defined subtype [1]. In most clinical practices, immunohistochemistry-based loss of SMARCB1/INI1 protein expression is used as a surrogate for pathogenic (biallelic) alterations of SMARCB1, which can occur through diverse mechanisms [1, 21, 24, 25]. The largest study to date evaluated 38 renal medullary carcinoma cases using FISH (for translocations and copy number alterations) and next generation sequencing (for copy number alterations, and single nucleotide variants/small insertion deletion events) [25]. The results showed that the most common event involved biallelic inactivation of the SMARCB1 tumor suppressor gene through hemizygous loss/ translocations (52.6%), followed by homozygous loss (15.8%), while less frequent were deletion of one SMARCB1 allele and an indel of the second SMARCB1 allele (13.2%), while deletion of one SMARCB1 allele and a truncating pathogenic alteration was identified in a single case (2.6%) [25]. A genetic mechanism of SMARCB1 loss could not be identified in 15.8% of cases and this may be secondary to epigenetic changes. These results validated the findings of two previous studies in a total of 25 patients with renal medullary carcinoma that noted hemizygous deletion/ translocations as the most common mechanism of SMARCB1 loss in 60% of cases [21, 24].
While the hypoxemic microenvironment of the renal medulla and SMARCB1 loss are central to the pathogenesis of renal medullary carcinoma [1, 4, 10, 21, 26], other oncogenic mechanisms continue to be identified. High iron concentrations within the renal medulla have been linked to ferroptosis, which is an iron-dependent mode of programmed cell death [27]. The thick ascending limb of the loop of Henle within the renal medulla is exposed to high osmolarity and hypoxemia, which is exacerbated in the setting of sickle cell trait, leading to iron release by sickled red cells [28]. A recent study utilizing single cell sequencing identified cells in the thick ascending limb as the putative cell of origin of renal medullary carcinoma, with ferroptosis resistance providing a selective pressure for these cells to lose SMARCB1 [28]. These results are consistent with a previous study of single-cell RNA sequencing in a single patient with medullary carcinoma, which suggested the thick ascending limb of the Loop of Henle within the renal medulla as the site of origin of renal medullary carcinoma [29].
HISTOPATHOLOGIC FEATURES
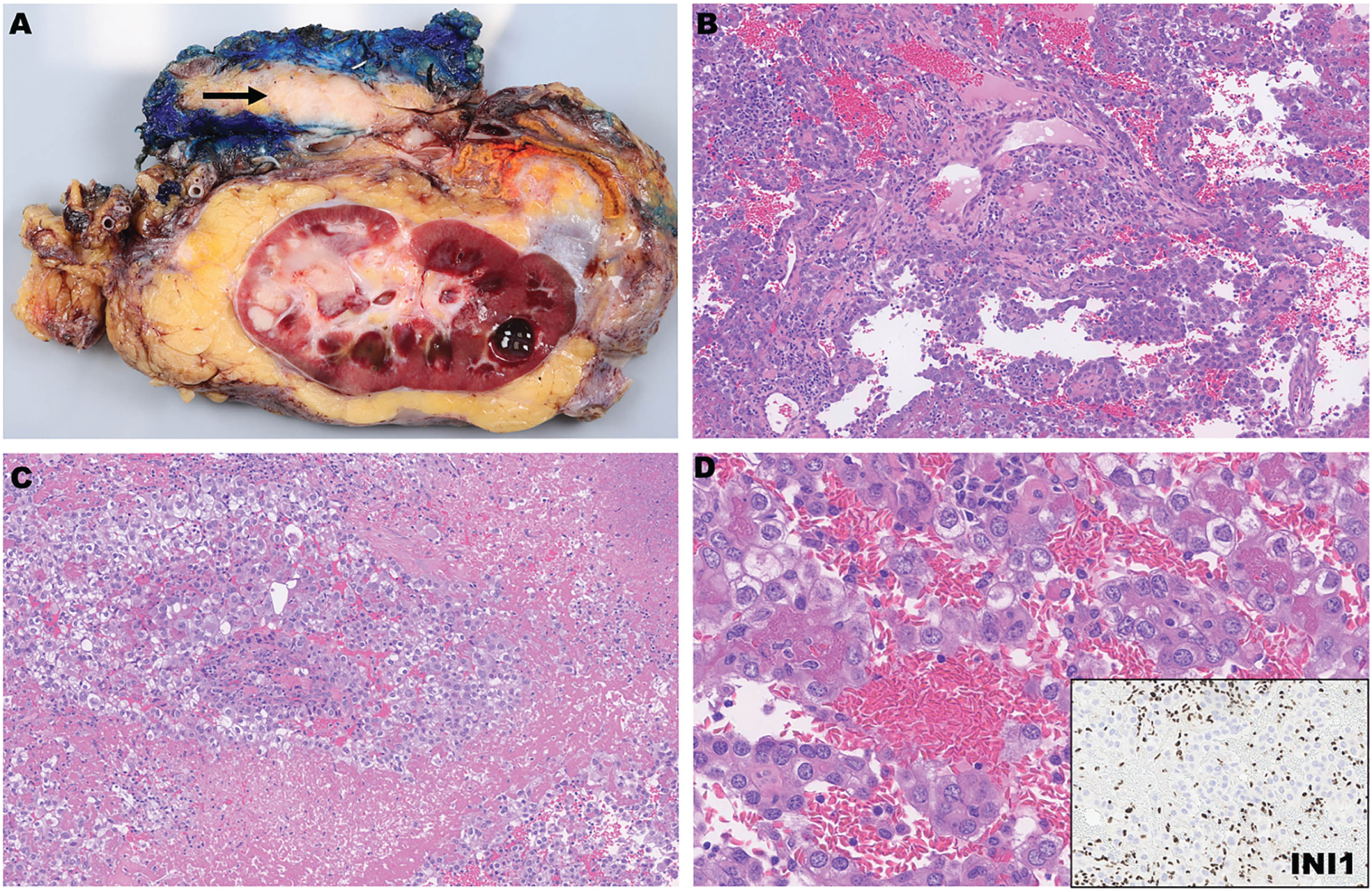
Renal medullary carcinoma typically arises from the renal medulla (Fig. 1A-B) but can originate from the renal cortex [1, 12, 13, 19], and these tumors have a reported median size of 6 to 7.9 cm [11, 13–15, 19, 30–32]. Aggressive features such as tumor necrosis (Fig. 1C), sarcomatoid transformation, extension into the renal sinus/perinephric fat, lymphovascular invasion, and metastatic disease are frequently identified [4, 11–15, 19, 31, 32]. On histologic examination, these tumors exhibit diverse morphologic features and proliferate predominantly as cribriform glands, solid sheets, cords, and nests with desmoplastic and or myxoid stroma [4, 11]. Occasionally rhabdoid morphology is seen, and sickled erythrocytes (drepanocytes) can usually be identified (Fig. 1D) [4, 11, 13]. On immunohistochemistry renal medullary carcinoma exhibits complete (nuclear) loss of SMARCB1/INI1 expression, with the adjacent immune cells and non-neoplastic elements serving as an internal control (Fig. 1D, inset) [4, 14, 19]. Additionally, a subset of renal medullary carcinoma characteristically expresses OCT3/4, which can be of use to separate it from other tumors which show overlapping morphologic features [15]. Specifically, positivity for OCT3/4 has been reported in 39% to 71% of cases, often with variability in extent of expression [4, 15, 21].
DIFFERENTIAL DIAGNOSIS OF RENAL MEDULLARY CARCINOMA
Renal medullary carcinoma shows significant morphologic overlap with several other distinct renal cell carcinoma subtypes. Relevant clinical features, immunohistochemistry, and identification of underlying molecular alterations can reliably separate these entities, and this has been discussed below. Some relevant diagnostic features are summarized in Table 1.
Differential diagnosis of renal medullary carcinoma.

Abbreviations. IHC: immunohistochemistry; FH: fumarate hydratase; 2SC: S-(2-succino)-cysteine; RCC: renal cell carcinoma; RMC: renal medullary carcinoma; MTSCC: mucinous tubular and spindle cell carcinoma. ||Hemoglobinopathies (HbAS: sickle cell trait; HbSC: hemoglobin SC disease; HbSS: sickle cell disease; HbS/ß°: hemoglobin S-beta thallasemia disease).
Collecting duct carcinoma
Collecting duct carcinoma, initially described by Fleming and Lewi as a distinct subtype of renal cell carcinoma in 1986, has undergone significant evolution in diagnostic criteria [4, 26, 33–35]. Morphologic features of collecting duct carcinoma include involvement of the renal medulla, cytologic atypia, tubular, and infiltrative growth, accompanying inflammatory infiltrates, and a desmoplastic stromal response [4, 26, 33, 34, 36]. However, in the absence of recurring molecular alterations this has become a diagnosis of exclusion [34]. Tumor types that must be excluded before making a diagnosis of collecting duct carcinoma include urothelial carcinoma, renal medullary carcinoma, fumarate hydratase-deficient renal cell carcinoma, high-grade mucinous tubular and spindle cell carcinoma, ALK-rearranged renal cell carcinoma, and SMARCB1-deficient medullary-like renal cell carcinoma [16, 34]. Of note, collecting duct carcinoma shows a high level of immunophenotypic overlap with upper tract urothelial carcinoma as both can express PAX8, and high molecular weight keratins [34].
Upper tract urothelial carcinoma
High grade invasive urothelial carcinoma can have a similar location in the kidney as collecting duct and renal medullary carcinoma and show significant morphologic overlap [34]. Furthermore, it is important to note that while PAX8 is considered a marker of renal histogenesis, its expression has been reported in a subset of upper tract urothelial carcinoma [33, 37, 38]. While markers such as high molecular weight keratins may be expressed by both urothelial and collecting duct carcinoma, other markers such as SMARCB1/INI1, P63 and GATA3 may be helpful in discriminating between these entities [34, 37, 38]. Newer diagnostic markers of urothelial lineage include uroplakin II and III, however, additional data is needed to understand the specificity of these markers for urothelial lineage [39]. Finally, detection of hotspot promoter mutations of the TERT gene is often used to establish urothelial lineage as it is present in over 70% of cases, however, this is unlikely to be helpful as such alterations are also present in a subset of renal cell carcinomas [40].
Fumarate hydratase-deficient renal cell carcinoma
Fumarate hydratase (FH) alterations are identified in a subset of renal cell carcinoma that can occur both in the germline (hereditary leiomyomatosis and renal cell cancer, HLRCC) [41] and sporadic setting [4, 16, 26]. These tumors can show significant morphologic overlap with renal medullary carcinoma, including sieve-like/ cribriform architecture [26]. The FH-deficient renal cell carcinoma nomenclature is preferred as germline sequencing results are often unavailable at the time of diagnosis [26]. In clinical practice, immunohistochemistry using a combination of FH and S-(2-succino)-cysteine (2SC) is used to screen for these tumors [42–44]. While most FH-deficient tumors show loss of FH coupled with aberrant overexpression of 2SC (FH–/2SC+ immunophenotype), in rare instances dysfunctional FH protein may be retained within the tumor cells leading to a FH+/2SC+ immunophenotype [44]. As opposed to molecular diagnostic tools, these immunohistochemistry-based surrogates are widely available and in a single multiinstitutional study, 25% of cases originally diagnosed as collecting duct carcinoma were reclassified as FH-deficient renal cell carcinoma [4].
High-grade mucinous tubular and spindle cell carcinoma
Mucinous tubular and spindle cell carcinoma can show significant morphologic and immunophenotypic overlap with papillary renal cell carcinoma. High-grade tumors with predominantly tubular morphology that infiltrate into the renal medulla can pose a diagnostic challenge. While these tumors do not have a very specific immunophenotype, they are characterized by distinct cytogenetic changes [45] as well as the overexpression of the VSTM2A gene, which can be identified using in-situ hybridization [46]. Characteristic copy number changes in these tumors include loss of chromosomes 1, 4, 6, 8, 9, 13, 14, 15, and 22, while additional alterations seen in tumors with high-grade transformation include homozygous loss of CDKN2A/B and gain of chromosome 1q [47].
ALK-rearranged renal cell carcinoma
Renal cell carcinomas with rearrangements of the ALK gene were included in the current WHO classification as a molecularly defined subtype of renal cell carcinoma. While ALK-rearranged renal cell carcinoma occurring in adults have a broad morphologic spectrum and gene fusion partners (CLIP1, EML4, HOOK1, KIAA1217, KIF5B, PLEKHA7, STRN, TPM3), they typically lack sickle cell trait [48]. In contrast, tumors reported in younger patients (6 to 22 years of age), are usually characterized by the presence of sickle cell trait and involve rearrangements with the VCL gene and these tumors have therefore been proposed as the “eighth sickle-cell nephropathy” [49–52]. Consequently, testing for ALK rearrangements is recommended in pediatric patients with renal cell carcinoma occurring in the setting of sickle cell trait that show retained expression of SMARCB1/INI1. Importantly, identification of ALK rearrangements may make these patients eligible for targeted therapy with agents such as entrectinib and crizotinib [51, 52].
SMARCB1-deficient renal medullary carcinoma & SMARCB1-deficient medullary-like renal cell carcinoma
While the majority of renal medullary carcinoma arise in the setting of an underlying sickle hemoglobinopathy (HbAS, HbSC, HbS/ß-thalassemia), rare cases have been identified in patients that lack sickle hemoglobinopathy [11, 24, 53–57]. For this reason, the current WHO classification recommends the dual nomenclature of SMARCB1-deficient renal medullary carcinoma and SMARCB1-deficient medullary-like renal cell carcinoma, when associated with and without an underlying sickle hemoglobinopathy, respectively [1]. Importantly, SMARCB1-deficient medullary-like renal cell carcinoma shows histologic features that are similar to SMARCB1-deficient renal medullary carcinoma, including SMARCB1/INI1 loss [1, 4, 33, 53, 58].
Renal cell carcinoma with secondary SMARCB1-loss
Finally, in rare instances SMARCB1/INI1 loss has been documented in other established subtypes of renal cell carcinoma [1]. This includes TFE3 gene-rearranged renal cell carcinoma [59], and FH-deficient renal cell carcinoma [60, 61] with concurrent loss of SMARCB1/INI1. The current recommendation from the WHO is to classify such tumors based on the primary tumor type, with “secondary SMARCB1 loss” [1]. Other rare reports include SMARCB1-deficient medullary-like renal cell carcinoma (negative for sickle hemoglobinopathy) in a patient with neurofibromatosis type 2 [62], and loss of SDHB in a patient with SMARCB1-deficient renal medullary carcinoma in the setting of sickle cell trait [61].
FH-deficient renal cell carcinoma with secondary SMARCB1 loss deserve further mention. Rare cases have revealed the presence of SMARCB1 loss by immunohistochemistry in the setting of an underlying pathogenic alteration of the FH gene, without an identifiable underlying genetic alteration of SMARCB1. It has been proposed that silencing of SMARCB1 gene expression in such cases may occur secondary to epigenetic mechanisms [61]. In this context it is important to note that studies by The Cancer Genome Atlas (TCGA) and others revealed that a subset of FH-deficient renal cell carcinoma exhibited a CpG island methylator phenotype (CIMP) [63–65]. This is hypothesized to be secondary to inhibition of ten-eleven translocation methylcytosine dioxygenases (TETs) that are involved in the removal of new aberrant CpG methylation, due to an intracellular accumulation of fumarate [64]. Future studies are needed to confirm whether secondary SMARCB1 loss in FH-deficient renal cell carcinoma occur due to epigenetic silencing as a part of the CIMP phenotype.
Rhabdoid tumor & rhabdoid tumor predisposition syndrome
Other SMARCB1/INI1-deficient renal tumors include rhabdoid tumor of the kidney, and this usually does not enter the differential diagnosis of renal medullary carcinoma as these tumors typically occur in infancy and early childhood [66]. Although the median age at diagnosis of rhabdoid tumor of the kidney in one study was reported to be 10.6 months, rare cases have been reported in older patients (>8 years) [66]. Recent studies suggest that while renal medullary carcinoma arises from the thick ascending limb (epithelial) cells [28, 29], rhabdoid tumor of the kidney is derived from neural crest-derived Schwann cells [67]. This is further supported by distinct methylation and transcriptomic profiles of renal medullary carcinoma compared to rhabdoid tumor of the kidney, where the former shows overexpression of gene such as FOXI1, which is known to regulate early nephrogenesis [68].
Other distinguishing features include only focal PAX8 positivity in rare cases of rhabdoid tumor of the kidney [69]. Furthermore, renal and extrarenal malignant rhabdoid tumors can occur in patients with SMARCB1 (type 1 rhabdoid tumor predisposition syndrome, >98% of cases) and SMARCA4 (type 2 rhabdoid tumor predisposition syndrome) germline alterations [70].
OUTCOMES AND MANAGEMENT OF SMARCB1-DEFICIENT RENAL MEDULLARY CARCINOMA
Patient outcomes
In one study, the median survival for patients diagnosed with renal medullary carcinoma was 7.7 months, and the 1-year, 3-year, and 5-year survival was 33%, 11%, and 9%, respectively [30]. More contemporary multicenter data suggest that this median survival may have improved to 13 months [6, 71]. Most patients (65–90%) present with metastatic disease at the time of diagnosis [1, 2, 11, 30, 31]. In patients with metastatic disease at presentation survival is estimated to be 4-5 months, as opposed to 17-18 months in patients without metastatic disease at presentation [30, 31]. Of note, patients with metastatic disease rarely survive longer than 3 years [30], while approximately 20% of patients without metastatic disease survived beyond 5 years [30]. This highlights the need for early recognition and management of renal medullary carcinoma [6].
Treatment of renal medullary carcinoma
The targeted therapies developed for the treatment of clear cell renal cell carcinoma, including anti-angiogenic tyrosine kinase inhibitors and MTOR inhibitors, are ineffective against renal medullary carcinoma and should be avoided [6, 71, 72]. Platinum-based cytotoxic chemotherapy such as carboplatin plus paclitaxel is typically used as the first line regimen both in patients with localized disease prior to nephrectomy, due to the high risk of metastasis, and in patients with metastatic disease [72, 73]. First-line platinum-based chemotherapy can produce typically brief responses in ∼29% of patients [71]. The combination of gemcitabine plus doxorubicin can be an effective second-line regimen that can produce responses in ∼19% of patients with platinum-refractory disease based on retrospective data in 16 patients [74]. A clinical trial (NCT03587662) was recently completed and will report the efficacy of adding the proteasome inhibitor ixazomib to the backbone of gemcitabine plus doxorubicin in 30 patients with renal medullary carcinoma [75].
Consistent with the significant upregulation of the EGFR pathway noted in renal medullary carcinoma tissues [25], retrospective data suggest that the combination of the EGFR inhibitor erlotinib with bevacizumab produced responses in ∼20% of heavily pre-treated patients with renal medullary carcinoma, most of whom had progressed on first-line platinum-based chemotherapy and second-line gemcitabine plus doxorubicin. However, recent preclinical data suggest that SMARCB1 loss protects renal medullary carcinoma from anti-angiogenic therapies with agents such as bevacizumab [22]. This is consistent with a recent phase 2 clinical trial where the addition of bevacizumab to platinum-based chemotherapy did not improve responses while increasing grade 3-4 toxicities in patients with renal medullary carcinoma [73].
Pre-clinical studies and clinical trials are underway to develop more effective therapeutic regimens for renal medullary carcinoma. Prospective clinical trials have now shown that immune checkpoint blockade with either pembrolizumab monotherapy [76], or a combination of nivolumab and ipilimumab (NCT03274258) [72] does not produce responses in patients with renal medullary carcinoma. Due to the significant upregulation of LAG3 in renal medullary carcinoma [25], an ongoing clinical trial is evaluating high doses of the LAG3 inhibitor relatlimab combined with nivolumab in patients with renal medullary carcinoma (NCT05347212). A recent preclinical study showed promising results when the neddylation pathway inhibitor pevonedistat was combined with platinum-based chemotherapy in both in-vitro and in patient-derived xenograft models of renal medullary carcinoma [77]. Inhibition of neddylation can aggravate both the proteotoxic and replication stress induced by SMARCB1 loss in renal medullary carcinoma leading to cell death [25, 75, 77].
SUMMARY
Recent studies have improved our understanding of the pathophysiology of renal medullary carcinoma including high-intensity exercise in patients with sickle cell trait as a potential modifiable risk factor, thick ascending limb cells as the putative cell of origin, and mechanisms of SMARCB1 loss. Furthermore, the SMARCB1-deficient phenotype likely serves as an adaptation to ferroptosis and the hypoxemic microenvironment induced by red blood cell sickling in the renal medulla of individuals with sickle hemoglobinopathies. Current diagnostic algorithms classify these tumors as SMARCB1-deficient renal medullary carcinoma, and SMARCB1-deficient medullary-like renal cell carcinoma based on the presence of sickle hemoglobinopathies, and established subtypes of renal cell carcinoma with secondary SMARCB1 loss continue to be identified. These algorithms help in separating the above-mentioned tumors from morphologic mimics which include collecting duct carcinoma, upper tract urothelial carcinoma, FH-deficient, and ALK-rearranged renal cell carcinoma which have different natural histories and therapeutic implications. Finally, there is an unmet need to develop better treatment regimens for patients with renal medullary carcinoma given its aggressive clinical course and several agents continue to be evaluated, in addition to standard-of-care cytotoxic chemotherapy regimens.
Footnotes
ACKNOWLEDGMENTS
Pavlos Msaouel is supported by the Andrew Sabin Family Foundation Fellowship, Gateway for Cancer Research, a Translational Research Partnership Award (KC200096P1) by the United States Department of Defense, an Advanced Discovery Award by the Kidney Cancer Association, a Translational Research Award by the V Foundation, the MD Anderson Physician-Scientist Award, donations from the Renal Medullary Carcinoma Research Foundation in honor of Ryse Williams, as well as philanthropic donations by the Chris “CJ” Johnson Foundation, and by the family of Mike and Mary Allen. The authors thank Brad Mosburg, PA (ASCP)CM, MHS, AAPA for his gross photography and detailed macroscopic findings.
FUNDING
The authors report no funding.
AUTHOR CONTRIBUTIONS
All authors participated in the conception, writing and reviewing of the manuscript.
CONFLICTS OF INTEREST
Pavlos Msaouel has received honoraria for service on a Scientific Advisory Board for Mirati Therapeutics, Bristol Myers Squibb, and Exelixis; consulting for Axiom Healthcare Strategies; non-branded educational programs supported by DAVA Oncology, Exelixis and Pfizer; and research funding for clinical trials from Takeda, Bristol Myers Squibb, and Mirati Therapeutics. Rumeal D. Whaley, John C. Cheville and Sounak Gupta have no relevant financial relationships with commercial interests to disclose.