
Abstract
Tumors with clear cells represent the most common group of renal neoplasms, with clear cell renal cell carcinoma (RCC) remaining the prototypical entity. Consequently, it is important to distinguish clear cell RCC from other entities with clear cell morphology, including TFE3-rearranged RCC, multiloculated cystic renal neoplasm of low malignant potential, clear cell (tubulo)papillary renal cell tumor, ELOC-mutated RCC, and RCC with fibromyomatous stroma. Some of these tumors can be aggressive, while others are almost always indolent. Accurate diagnoses prevent indolent cases from receiving unnecessary therapy or from inappropriately being enrolled in clinical trials with the potential of confounding their results. Molecularly classifying tumors may also lead to drugs that selectively target an oncogenic driver, thereby more effectively targeting the tumor. In current clinical practice, high-quality immunohistochemistry and molecular assays are continually enhancing our diagnostic ability. A discussion of these histotypes with clear cell morphology and how to discriminate them is the focus of this review.
INTRODUCTION
Renal tumors have traditionally been classified based on morphology and immunohistochemistry (IHC) [1]. Although morphology remains imperative, recent years have ushered in a molecular revolution in renal tumor classification [2–4]. For instance, a new category of molecularly defined renal cell carcinoma (RCC) now exists, containing some entities in the differential of renal tumors with clear cells, including TFE3-rearranged RCC and ELOC (formerly TCEB1)-mutated RCC [3, 5, 6]. Clear cell RCC (CCRCC) remains the most common subtype of RCC [7]. Other neoplasms have been deemed indolent, including multiloculated cystic renal neoplasm of low malignant potential (MLCNLMP) and clear cell (tubulo)papillary renal cell tumor (formerly clear cell (tubulo)papillary renal cell carcinoma) (CCTPRCT) [7]. The last entity to be considered, RCC with fibromyomatous stroma (FMS), is still being fully characterized at the molecular level [5]. In addition to the above entities that will be discussed in detail, several other neoplasms can have clear cells and enter the differential diagnosis when a kidney tumor with clear cells is encountered, including but not limited to epithelioid angiomyolipoma, TFE3-rearranged epithelioid angiomyolipoma, and adrenal cortical carcinoma. However, these lesions are outside the scope of the current review.
CLEAR CELL RENAL CELL CARCINOMA (CCRCC)
Clinical
CCRCC remains the most common type of RCC, accounting for 65–70% of cases [8]. The median age is in the seventh decade, but CCRCC still accounts for 54% of tumors in patients 20–39 yo [7, 9]. CCRCC is more common in men (2 : 1) and more common in Caucasians [7]. Risk factors include smoking, obesity, long-term dialysis, hypertension, diabetes mellitus and certain environmental exposures [7].
Less than 5% of cases are multifocal or bilateral [7]. Many cases are incidentally detected on imaging, but may present with back pain, weight loss, or hematuria [7]. A subset of cases first present with metastases due to a propensity to spread through the blood, which is heavily related to the grade [7, 10].
Tumor grade strongly impacts the risk of recurrence, with estimated probabilities within 5 years of follow-up being 0%, 7%, 22% and 52% for grades 1, 2, 3, and 4 respectively [11]. Tumors with sarcomatoid differentiation have a particularly poor outcome, with a 15–22% 5-year survival [7]. However, recent trials have shown benefit with immunotherapy in CCRCC with sarcomatoid differentiation [12, 13]. Stage at presentation and necrosis are further predictors of outcome [7].
The characteristic molecular alteration in CRCC is alteration of the VHL gene (loss of chromosome 3p) [8]. However, CCRCC may possess several additional molecular alterations, most commonly BAP1, SETD2, and PBRM1. CCRCC with BAP1 mutations behave more aggressively, while PBRM1 mutated tumors are thought to act more favorably [2, 14]. For instance, in one study, approximately two-thirds of BAP1-mutated CCRCCs were stage pT3 and 50% developed metastases [2]. SETD2 mutated tumors may have a worse prognosis, but some studies have found less influence on prognosis [15–17]. However, these markers are not in routine use in all laboratories for prognostic stratification and are not included in current clinical guidelines [2]. However, they may have emerging roles as our availability and integration of molecular data evolves.
These novel molecular findings may soon have therapeutic implications as well. For example, CCRCC with a strong angiogenic gene signature had a favorable response to angiogenic therapies and were enriched for PBRM1 loss [7]. The loss of BAP1 results in accumulation of ubiquitinated histone H2A, for which histone deacetylase inhibitors could be beneficial [2]. In addition, the polyADP ribose polymerase (PARP) inhibitor olaparib has been shown to be useful in BAP1-mutated cancer cells and may be useful in BAP1 mutated tumors, but further investigation is needed [2, 18].
Gross and microscopic findings
The mean size is 6.3 cm (range 1.2–17) [7]. Tumors usually have a round, pushing expansile appearance with golden-yellow cut surfaces with frequent hemorrhage. Cystic change is frequent in low grade tumors, while high-grade tumors may possess whitish, firm areas with frequent necrosis, often with extension into the renal sinus [7].
Microscopically, tumors characteristically have a nested, tubular, or alveolar growth pattern comprised of cells with clear cytoplasm (Fig. 1A) [7]. However, morphological variation is common, such as cystic change, degenerative changes, or hemorrhage. There is a complex vascular network with capillaries surrounding each nest of tumor cells. Higher grade tumors often have eosinophilic cells or hyaline globules. Giant multinucleated cells, sarcomatoid, or rhabdoid features can also be present [7].
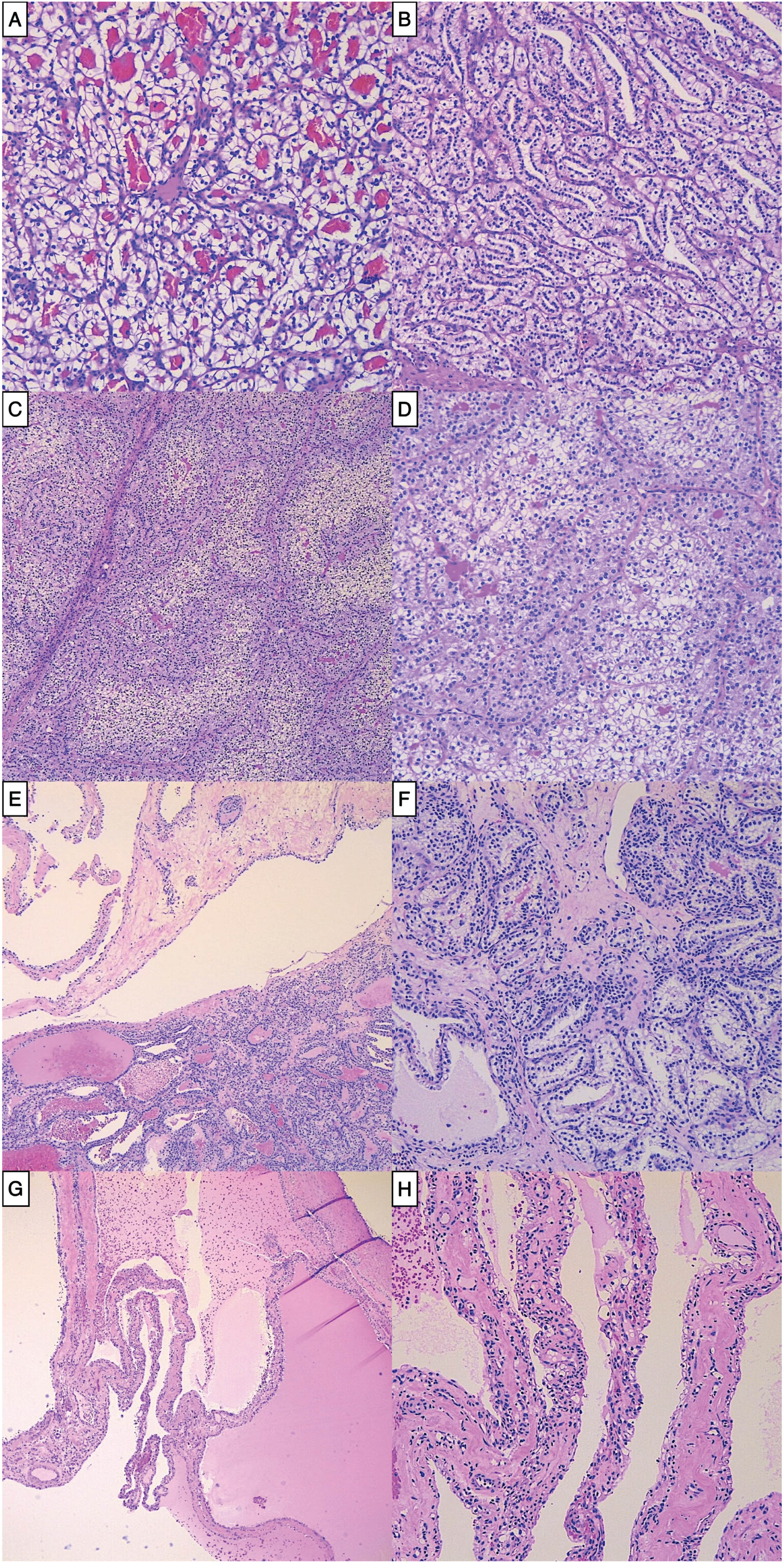
A. Characteristic CCRCC with nests of clear cells (100x); B. CCRCC showing nuclei lined up away from the basement membrane, mimicking CCTPRCT (100x). C. Low power image of TFE3 RCC displaying two-toned cytoplasm (40x). D. TFE3 RCC showing a mix of eosinophilic and clear cells with voluminous cytoplasm (100x). E. CCTPRCT with cystic and solid areas (40x). F. CCTPRCT showing tubules of low-grade clear cells with lined up nuclei away from the basement membrane (100x). G. MLCNLMP at low power (20x). H. MLCNLMP with single layers of bland clear cells lining septae (100x).
Ancillary studies and molecular alterations
Carbonic anhydrase IX (CA9) is a hypoxia driven marker related to VHL and is diffusely positive circumferentially (box-like) [19]. CK7 is often negative or focal but can be more strongly positive in cystic areas [8]. CD10 is usually positive, while AMACR is variable and can be negative [7, 20].
CCRCC harbors alterations of the VHL gene (loss of chromosome 3p), either in the form of mutation or promoter methylation and a second hit typically occurs as a large deletion [8]. VHL is part of a stable complex that targets hypoxia-inducible factor alpha (HIFa) subunits for degradation. The lack of functional VHL stabilizes HIFa, which leads to downstream angiogenesis and cell proliferation [21]. A VHL gene alteration can also serve as a potential diagnostic marker, as it can be detected by FISH or chromosomal microarray studies [7, 8]. Additional mutations in tumor suppressor genes have also been detected, including PBRM1 (∼40–50%), SETD2 (∼10–20%), and BAP1 (∼12%-15%) [2, 8].
Most CCRCC are sporadic, but may be familial, including in von Hippel-Lindau syndrome, BAP1 tumor disposition syndrome, Cowden syndrome, Birt-Hogg-Dube syndrome, and tuberous sclerosis (TSC) [7].
Differential diagnosis
The differential diagnosis for CCRCC includes CCTPRCT, TFE3 RCC, chromophobe RCC (ChRCC), MLCNLMP, ELOC-mutated RCC, and RCC FMS (Table 1) [7]. The distinction from CCTPRCT and MLCNLMP is critical as these two entities are not classified as carcinomas, while RCC FMS and ELOC-mutated RCC are relatively indolent compared to CCRCC [22]. Clinical trials are often still separated based on CCRCC vs non-CCRCC, further emphasizing the importance of their distinction [23].
Comparison of renal cell tumors with predominantly clear cell morphology.
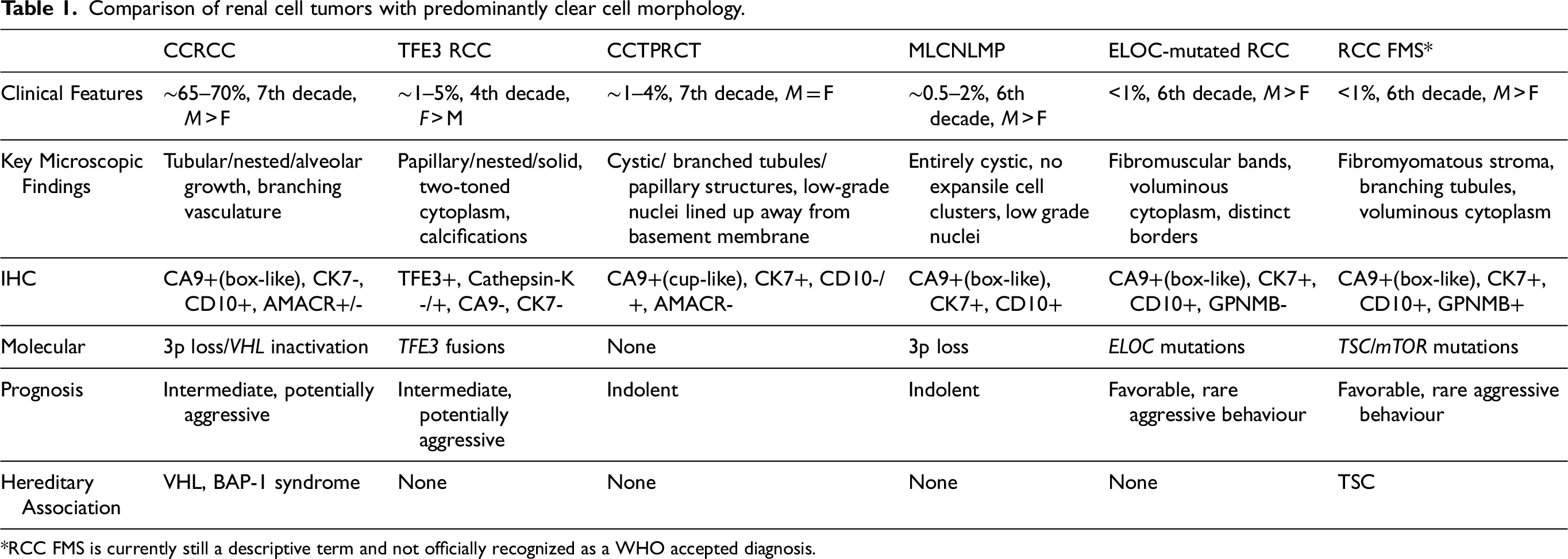
*RCC FMS is currently still a descriptive term and not officially recognized as a WHO accepted diagnosis.
TFE3 RCC
This is often the most difficult distinction in the appropriate clinical setting [2, 24]. Younger patient age, pale to eosinophilic voluminous cytoplasm, papillary architecture, and psammomatous calcifications favor TFE3 RCC [25]. Conversely, pseudopapillary architecture, extreme nuclear pleomorphism, rhabdoid, or sarcomatoid differentiation favor CCRCC [25]. Nevertheless, the two can show significant morphologic overlap, necessitating IHC and/or molecular studies [26]. Diffuse, strong TFE3 IHC is the most confirmatory biomarker for TFE3 RCC [27, 28]. If TFE3 is unavailable, CA9 is diffusely and strongly positive in CCRCC but negative or only focally positive in most TFE3 RCC [19, 25, 26]. If positive, cathepsin-K is also helpful, as it is negative in CCRCC [27, 29]. Lastly, TFE3 RCC can under express epithelial markers and occasionally expression melanocytic markers[25, 30]. If ambiguity remains, cytogenetics or FISH can be employed, with CCRCC having 3p deletion and lacking TFE3 rearrangements [26, 27].
CCTPRCT
Higher grade areas, necrosis, or regional invasion will exclude the diagnosis of CCTPRCT [31, 32]. However, tumors may be small, confined, low grade, and even have branched glandular structures, prominent papillary structures, and nuclear alignment closely mimicking CCTPRCT (Fig. 1B) [7, 33]. Moreover, small sampling in core biopsies may make the distinction based on morphology alone impossible [34]. Indeed, CCTPRCT-like morphology can occupy up to 90% of a CCRCC on resection, and thus be entirely CCTPRCT-like on biopsy[35]. Utilizing IHC will lead to the correct diagnosis in most cases, as the CA9 staining pattern will differ (box-like in CCRCC vs cup-like in CCTPRCT) [7, 31]. CK7 will be diffusely positive, while AMACR and CD10 will be negative in CCTPRCT, all three of which will have the opposite pattern in CCRCC [7, 31]. However, on small core biopsies, even IHC may not distinguish the two entities. In these rare instances that remain equivocal, molecular studies may be necessary, in which VHL mutation or chromosome 3p loss will confirm the diagnosis of CCRCC [8, 32, 33].
ChRCC
The differentiation from ChRCC is usually straight forward on morphology. However, in small samples, CCRCC can be rather eosinophilic and ChRCC can be quite clear. In these cases, IHC will solve the dilemma as CCRCC is diffusely positive for CA9, while ChRCC is uniformly negative [19]. In addition, ChRCC will be positive for CK7 and CD117, while CCRCC will be the opposite [36].
ELOC-mutated RCC
It is very difficult to discriminate ELOC-mutated RCC from CCRCC [8]. Tumors with ELOC mutations also have dysregulation of the VHL pathway, resulting in overlapping morphology and IHC profile, such as diffuse positivity for CA9 [7, 8]. An initial clue can be the multi-nodular appearance of ELOC-mutated RCC at low power [37]. Branching convoluted tubules and papillae are also more common in ELOC-mutated RCC [37]. An additional clue would be diffuse labelling for CK7 in ELOC-mutated RCC, although this is not specific enough for a definite diagnosis without further confirmation, making the demonstration of biallelic ELOC alterations a requirement for diagnosis [3, 8]. Conversely, tumors with 3p deletion or monoallelic ELOC deletion would be diagnosed as CCRCC [3].
MLCNLMP
The distinction may not be possible on core biopsy, as MLCNLMP cannot be diagnosed on a small sample and the IHC profile is similar. On resection, small, solid, expansive nodules (exceeding 1 mm in size) or necrosis excludes the diagnosis of MLCNLMP [7].
RCC FMS
CCRCC usually lacks fibromyomatous stroma and papillary structures, both of which are often present in RCC FMS [22]. However, these two entities have overlapping morphology and IHC profiles, making the distinction without molecular confirmation difficult. VHL inactivation is characteristic of CCRCC, while RCC FMS contains TSC1/2, mTOR, or ELOC alterations [22].
TFE3 REARRANGED RENAL CELL CARCINOMA (TFE3 RCC)
Clinical
Overall, TFE3 RCC is relatively uncommon, accounting for 20–70% of pediatric RCC [38–40], up to 15% in adults under age 45, and < 5% (range 0.2 – 5%) of adult RCC [23, 27, 41–45]. The mean age is in the fourth decade (range 0–84 yo) [27, 46–48], but quite rare in males over 60 yo [25, 47, 49–54]. Tumors are more common in females, with a female to male ratio up to 6 : 1 [27, 39, 55].
Although studies have found mixed results, TFE3 RCC in children tends to have a more indolent course [23, 50, 56–60]. Conversely, TFE3 RCC in adults is more aggressive than CCRCC, frequently presenting with locally advanced or metastatic disease [27, 49, 61, 62]. There is lymph node involvement at the time of diagnosis in 26 to 84% and distant metastases in 14 to 35% of cases [27, 60, 63, 64]. Stage 1 and 2 cases largely have good outcomes, with only rare recurrences [47]. Patients with at least stage pT3a have had recurrence rates from 85 to 100% and mortality of 45% to 57% [25, 47, 59]. In addition to stage, older age has been reported as a significant unfavorable prognostic factor in multivariate analysis [27, 47, 59, 65].
It has not been firmly established whether biologic potentials differ for the various fusion types [27, 63]. Overall, the data is too scarce and the follow-up too short to conclude any definite connection independent of advanced age and stage [47, 63, 66]. However, one of the most common fusion partners, ASPSCR1, appears to be have the worst prognosis [27].
To date, there are no treatment recommendations specific for TFE3 RCC [23, 67, 68]. Current clinical trials predominantly focus on tumors with clear cell histology [23]. For advanced stage disease, immunotherapy and targeted therapies (anti-VEGFR [vascular endothelial growth factor receptor], mTOR inhibitors, MET tyrosine kinase inhibitors) can be used [23, 69]. Immune checkpoint inhibitors (ICI) appear to be more effective when compared to tyrosine kinase inhibitors (TKI) [6, 23, 67]. However, responses have still been relatively poor. One retrospective study reported overall response rates to dual ICI in only 5.5% of 18 patients with TFE3 RCC [69]. Important information could come from the only prospective clinical trial dedicated to TFE3 RCC patients, which has closed to accrual but still ongoing [67]. Moving past currently available drugs, the ideal therapy for these tumors may be a molecule that directly targets the fusion [67].
Gross and microscopic findings
Grossly, thick fibrous capsules or pseudocapsules may be present [46, 70]. The cut surface is typically variegated tan-yellow/brown and solid with frequently admixed hemorrhagic or necrotic foci [27, 29, 52]. Mean tumor sizes have been reported from 5.2 to 12.5 cm (range 0.8–25) [25, 46, 48, 49, 70–73].
Tumors are architecturally and cytologically heterogeneous [27, 74]. Architecturally, they often contain a mix of papillary, nested, solid, and cystic areas [27, 49, 73]. Tumors are composed of large polygonal cells with discrete borders, voluminous granular eosinophilic to clear cytoplasm (often dual toned) (Fig. 1C, D), vesicular nuclear chromatin, and hyaline nodules [7, 27, 49, 73]. Psammomatous calcifications are a sensitive, but non-specific finding [27, 73].
Ancillary studies and molecular alterations
IHC staining with the antibody from the C-terminal portion of TFE3 is the most commonly used, most sensitive, and most specific marker for TFE3 RCC [27]. However, TFE3 IHC is subject to technical difficulties, affected by fixation times, and lacks standardized interpretation guidelines, which can lead to false negative or positive results [23, 48–50]. In addition, staining can be equivocal and difficult to interpret [75]. Nevertheless, due to its rapidity and cost effectiveness, TFE3 IHC can be used as a screening method, followed by ISH or FISH in equivocal cases [49, 59].
CA9 is the second most useful IHC marker, being either negative or only focally positive in the large majority of cases, but has been reported in up to 37% of cases strongly in one study [19, 27, 28, 30]. Cathepsin-K is positive in a subset of cases, showing variability depending on the fusion partner, with an overall expression of 35% –60% in non-selected cases [25, 27, 29, 76]. With fusions known to be positive, rates are 60–100% [29, 47, 52, 71, 76], while as low as 0% if known to be negative [29, 47, 66, 71, 76]. EMA and pankeratin are under expressed, while CK7 is usually negative [25, 27, 30, 57]. HMB-45 and Melan-A are usually negative [77].
TFE3 break-apart fluorescent in situ hybridization (FISH) is currently the gold standard [23, 27]. Unfortunately, FISH has several technical issues related to fixation and cutting artifacts, as well as other deficiencies limiting its sensitivity, diagnostic accuracy, and uniform interpretation [27]. For instance, some fusion partners have easily recognized split signals [78, 79]. In contrast, some fusions caused by an inversion of chromosome X result in a short distance between the sequences targeted and a lack of split signals, thus being difficult to detect, specifically NONO, RBM10, and GRIPAP1.
Recently, RNA-ISH for TRIM63 has been identified as a sensitive and specific biomarker for TFE3 RCC [82]. When compared to FISH, TRIM63 mRNA was highly expressed in all cases of TFE3 RCC compared to other subtypes. Importantly, TRIM63 RNA-ISH was strongly positive in TFE3 FISH false-negative cases [82].
Many of the aforementioned deficiencies could be avoided by RNA-sequencing and RT-PCR, which has accelerated several areas in pathology including TFE3 RCC identification [27, 41]. However, RT-PCR is difficult to implement since it is not readily applicable to FFPE tissue due to inadequate RNA quality and can only identify known fusion variants [50, 71, 81]. Conversely, RNA-sequencing can simultaneously detect multiple gene rearrangements (without prior knowledge of fusion partners) with low quantities of tissue [27]. Indeed, RNA-sequencing has expanded the spectrum in recent years, with 22 genes now identified as fusion partners and 50% of novel fusion sub-types being identified since 2017 [27, 83]. ASPSCR1, PRCC, and SFPQ1 remain the most common partners [27].
Differential diagnosis
The wide morphologic spectrum necessitates consideration not only with tumors possessing clear cell and/or papillary features, but also unusual or difficult to classify RCC [26]. However, the differential primarily includes CCRCC, Papillary RCC (PRCC), CCTPRCT, MLCNLMP, and ELOC-mutated RCC [26, 27, 84]. The distinction from CCRCC has been previously discussed.
PRCC
When PRCC has clear cell features, the overlapping morphology often necessitates ancillary studies [26]. The absence of CK7, pankeratin, and EMA, while positivity for cathepsin-K and TFE3 favors TFE3 RCC [26, 27, 29]. If necessary, PRCC will show trisomy 7 and 17, while TFE3 RCC will have TFE3 gene rearrangements [26].
CCTPRCT
The subnuclear clearing characteristic of CCTPRCT can be present in TFE3 RCC [66, 73]. In particular, it has been recently noted that TFE3 RCC with the fusion partner NONO has nuclear alignment mimicking that of CCTPRCT [32]. Deceptively, FISH may be falsely negative with this fusion partner [66]. IHC should still be helpful, as CCTPRCT stains diffusely with CA9 (cup-like) and CK7 [7]. Conversely, TFE3 RCC usually shows only focal CA9 (box-like) and is usually negative for CK7 [66]. Strong immunostaining of TFE3 would also discriminate the two [66].
MLCNLMP
TFE3 RCC can closely mimic MLCNLMP depending on the fusion partner. The fusion partner MED15 leads to cystic masses in 90% of cases, some of which are almost entirely so [84, 85]. Unlike usual TFE3 RCC, the cyst lining cells in MED15 cases can have nuclei with inconspicuous nucleoli [84]. However, focally there are usually small mass-forming aggregates between septae associated with psammomatous calcifications, arguing against the diagnosis of MLCNLMP [84]. Again, IHC will be helpful, with MLCNLMP being CA9 positive and TFE3 negative, opposite to TFE3 RCC [7, 84].
ELOC-mutated RCC
ELOC-mutated RCC is characterized by thick fibromuscular stromal bands, cells containing clear or granular cytoplasm, and tubular and/or papillary architecture [37]. Distinction from TFE3 RCC can generally be made based on TFE3 of CA9 staining, as the former is negative and the latter typically diffusely positive in ELOC-mutated RCC [37]. However, cytogenetic analysis for monosomy 8 is required for confirmation [5].
CLEAR CELL (TUBULO)PAPILLARY RENAL CELL TUMOR (CCTPRCT)
Clinical
CCTPRCT makes up as much as 1–4% of renal tumors [7, 86–88], historically most often mistaken for CCRCC [8]. The mean age is in the seventh decade (range 8–88 yo) with no gender predilection [7]. The vast majority are incidentally found [7]. Although first described in end-stage kidneys, the majority are found in healthy kidneys [89]. Multifocal and bilateral renal tumors occur in 20% and 25% of cases respectively [7, 89]. However, 50% of other tumors are a different subtype, making recognition important for follow up [7, 86].
CCTPRCT is an indolent neoplasm, with multiple studies demonstrating no recurrence or disease progression during follow-up [7, 86, 87, 89]. Consequently, CCTPRCT is no longer considered a carcinoma.
Gross and microscopic findings
Tumors are usually small (mean 1.8 cm, range 0.6–6.5), encapsulated, and well circumscribed [7, 31, 87]. Cystic change is very common [31]. Microscopically, they are characterized by branched glandular structures, low grade nuclei aligned above the basement membrane, and variable papillary structures protruding into cystic spaces (Fig. 1E, F) [8, 31, 33]. There is no invasion of vessels, renal sinus, or peripheral fat [7].
Ancillary studies and molecular alterations
CCTPRCT has a characteristic staining pattern with diffuse CK7 positivity, diffuse CA9 in a cup-shaped pattern, negative AMACR, and negative or focal weak expression of CD10 [8]. GATA3 is variably used and variably positive [31].
These tumors do not have a defining pattern of recurrent genetic copy number changes and no disease-specific genetic abnormalities that indicate a driver [7, 8]. Recent work has found depletion of mitochondrial DNA and resultant oxidative stress [32, 86].
Differential diagnosis
The differential includes CCRCC, TFE3 RCC, PRCC, MLCNLMP, ELOC-mutated RCC, and RCC FMS [33, 87]. The distinction from CCRCC and TFE3 RCC have been discussed above.
Papillary RCC
PRCC can have prominent clear cell changes and low-grade cytology [32]. However, PRCC with clear cells usually contains other characteristic features, such as foamy macrophages or psammoma bodies. In contrast to CCTPRCT, PRCC is always strongly positive for AMACR and CA9 is usually focal, as opposed to negative AMACR and diffuse CA9 in CCTPRCT [32].
MLCNLMP
As the majority of CCTPRCT are cystic, MLCNLMP enters the differential [31]. In addition, both tumors are lined by clear cells with bland cytology. The distinction relies on identifying solid areas, as any expansile growth confirmed microscopically excludes the diagnosis of MLCNLMP [31]. In addition, MLCNLMP will have a box-shaped CA9 expression pattern and positivity for CD10, while CCTPRCT will have cup-shaped CA9 and negative CD10 [7].
ELOC-mutated RCC and RCC FMS
ELOC-mutated and RCC FMS also expresses CK7 and CA9 [31]. However, both entities often have morphologic features rarely seen in CCTPRCT, including voluminous cells and high-grade nuclei [31]. Conversely, subnuclear vacuolization characteristic of CCTPRCT is usually absent in ELOC-mutated RCC and rarely seen in RCC FMS [3, 22]. Moreover, CD10 is usually positive in both, while negative in CCTPRCT [3, 32]. The CA9 staining pattern also differs, being box-like in these two tumors vs cup-like in CCTPRCT [37, 90]. If necessary, molecular studies would reveal an absence of mutations in TSC1/2, mTOR or ELOC in CCTPRCT [7, 22].
MULTILOCULAR CYSTIC RENAL NEOPLASM OF LOW MALIGNANT POTENTIAL (MCLNLMP)
Clinical
MLCNLMP accounts for 0.5–2.5% of renal tumors [7]. The median age is 55 yo (range 18–84) and there is a male predominance [7]. The vast majority are incidentally discovered, while concurrent CCRCC has been found in 5–13% of cases [7]. MLCNLMP has excellent outcomes regardless of tumor size, with no reports of progression or metastases with long term follow up [7].
Gross and microscopic findings
Tumors are well defined with a mean size of 3.9 cm (range 0.6–15) [7]. Microscopically, MLCNLMP is composed entirely of variably sized cysts filled with serous or hemorrhagic fluid (Fig. 1G) [7]. Cyst septa lack solid areas or necrosis, lined only by one or a few layers of clear, low-grade cells (Fig. 1H). Clear cell clusters may exist, but do not alter the septa contours or exceed a 20x (1 mm) field [7].
Ancillary studies and molecular alterations
MLCNLMP are diffusely positive for CA9, while often positive for CK7 [7, 91]. Tumors contain VHL gene alterations, with loss of the entire chromosome 3 or its short arm detected by FISH in 77% of cases, or inactivating VHL mutations in 25–40% [7]. Next generation sequencing has also shown a significant number of overlapping genes in both MCNLMP and CCRCC, with a lower mutation frequency in MCNLMP [7].
Differential diagnosis
The diagnosis of MLCNLMP cannot be made on biopsy alone because exclusionary features cannot be evaluated. On resection, the differential includes cystic CCRCC, CCTPRCT, and cystic TFE3 RCC [7]. The key differentiating features have been discussed above.
ELONGIN C MUTATED RENAL CELL CARCINOMA (ELOC-MUTATED RCC)
Clinical
ELOC-mutated RCC has a male predominance and mean age of 60 yo (range 38 to 78) [3, 92]. Due to the molecular confirmation requirement for diagnosis, they are invariably under diagnosed, with less than 50 cases reported [7]. Owing to its novelty and rarity, the prognosis is not confidently known [3]. Most cases have been low stage, low grade, and behaved indolently [3, 6, 8, 92]. However, rare cases have acted aggressively [4, 93].
Gross and microscopic findings
ELOC-mutated RCC is typically small and well-delineated, grayish yellow or brown, and 2–3.5 cm [3, 7, 92]. Tumors have a nodular appearance due to thick fibromuscular bands transecting the tumor, along with clear cells, voluminous cytoplasm, and prominent cell borders [3, 4, 37]. Well-formed papillae, solid alveolar architecture, branching or tubular structures, and cystic features are present in varying amounts [3, 7, 37]. The grade is usually low [37, 92].
Ancillary studies and molecular alterations
Tumors are usually positive for CA9, CK7, and CD10, while negative for TFE3 [3].
These neoplasms have mutations in the ELOC (TCEB1) gene at 8q21.11, resulting in biallelic inactivation of ELOC (ELOC mutation in combination with loss of the other allele, ELOC deletions on chromosome 8q, typically by the loss of the entire chromosome 8) [4, 7]. ELOC codes for Elongin C, a binding partner of VHL in the E3-ubiquitin ligase complex [3]. ELOC mutations result in non-binding with the VHL complex, thus preventing HIF degradation, resulting in similar downstream effects as the tumorigenesis pathway in CCRCC [3, 7, 67]. The similar downstream alterations result in overlapping morphology and IHC staining, necessitating molecular confirmation for definitive diagnosis [67]. Importantly, there is an absence of VHL, TSC1, TSC2, or mTOR inactivation [3].
Differential diagnosis
ELOC-mutated RCC can morphologically mimic CCRCC, TFE3 RCC, CCTPRCT, and RCC FMS [3]. The former three have been discussed above.
RCC FMS
GPNMB has recently been shown to be uniformly positive in RCC FMS and negative in ELOC-mutated RCC [94]. Aside from the promise of this new marker, morphology and IHC cannot provide a definitive distinction between RCC FMS and ELOC-mutated RCC, necessitating molecular studies showing a lack of TSC/mTOR mutations and biallelic ELOC loss in ELOC-mutated RCC [5]. Unfortunately, such analyses remain out of reach for many laboratories [5]. In addition, more work is needed to fully characterise the spectrum of RCC FMS that also include those with monoallelic ELOC-mutations [5]. Since both entities appear to have indolent behavior, the routine use of expensive sequencing to distinguish these two entities is not currently employed in all cases [22]. The future development of less expensive genetic profiling assays could justify subsequent ELOC, TSC/mTOR, and/or VHL mutation analysis of these tumors more routinely [3].
RENAL CELL CARCINOMA WITH FIBROMYOMATOUS STROMA (RCC FMS)
Clinical
RCC FMS has been under recognized and called by various names, making a true determination of its incidence difficult [1]. Of the known cases, the mean age is 57 yo (range 29 to 78) with a male predominance and solitary tumors [22]. Based on limited follow up, RCC FMS has shown a favorable prognosis [1]. However, a couple of cases have behaved more aggressively with distant metastases [22, 95].
Gross and microscopic findings
RCC FMS have a mean size of 2.3 cm (range 0.8 – 5.0) [22]. Tumors are tan/yellow to red with a pseudocapsule [22]. Originally described as RCC with leiomyomatous stroma, RCC FMS contains variable amounts of fibromyomatous stroma surrounding nodules of elongated and frequently branching tubules lined by cells with voluminous clear cytoplasm [1, 22]. Focal prominent papillary architecture and cytoplasmic eosinophilic globules may be present [1, 22]. Nuclei are ISUP/WHO grade 2 or 3 and 97% of cases are pT1a [22, 90].
Ancillary studies and molecular alterations
Tumors are positive for CK7, CD10, and CA9 (usually box-like) [1, 22]. Phospho-S6 ribosomal protein (pS6) expression, suggestive of a hyperactive mTOR pathway, has been reported [57]. Very recently, a promising IHC marker has been reported, with GPNMB being positive in all cases [94].
RCC FMS has recurrent mutations involving the TSC/mTOR pathway [3]. Rare cases have also shown concurrent ELOC mutations, but no VHL mutations [1, 22]. Although more commonly sporadic, identical tumors have been found in patients with TSC, suggesting the existence of hereditary and sporadic cases [3].
Differential diagnosis
RCC FMS remains a descriptive term and is not yet an accepted WHO diagnosis. Therefore, RCC FMS can have overlapping morphologies with CCRCC, CCTPRCT, and ELOC-mutated RCC [3], the comparisons of which have been discussed above. IHC can occasionally be helpful, but molecular is needed to establish a definitive diagnosis.
CONCLUSION
Renal cell tumors with clear cells often have incredibly overlapping morphologies but widely different prognoses. Cases can frequently be distinguished with commonly available ancillary techniques, while some still require molecular confirmation with more novel, advanced assays. In addition, future work will focus on subclassification of molecularly well-studied subtypes, such as CCRCC, and whether therapeutic implications can be derived from such studies.