
Abstract
Papillary renal cell carcinoma (pRCC) represents the second most common renal cell carcinoma. Diagnosis of pRCC has been challenged by emergence of several novel renal tumor entities with papillary morphology. Some of these have been found to constitute independent entities with specific molecular drivers, e.g. fumarate hydratase deficient renal cell carcinoma or TFE3-rearranged or TFEB-altered RCC. Others are still considered emerging entities, such as papillary renal neoplasm with reverse polarity associated with KRAS mutations and biphasic hyalinizing psammomatous RCC associated with NF2 mutations. The recognition of these entities has changed the spectrum of “true” pRCC, and subclassification into type 1 and type 2 is no longer recommended. With the discovery of novel entities with specific diagnostic molecular traits, it is expected that the spectrum of pRCC is becoming increasingly narrow. This will potentially allow for a better prediction of prognosis and treatment response of pRCC. In this review we discuss recent developments in the diagnosis of renal tumors with papillary morphology, including emerging/provisional papillary renal tumor entities. The evolution of renal tumor classification has improved our understanding of biologic behavior and molecular background of pRCC with consequences on histopathological and molecular pathological approaches.
EVOLUTION OF PAPILLARY RENAL CARCINOMA CLASSIFICATION
Papillary renal cell carcinoma (pRCC) represents the second most common type of renal cell carcinoma (RCC) after clear cell renal cell carcinoma (ccRCC), building up to 10-20% of renal epithelial tumors [1, 2]. They may be multifocal and bilateral, especially in the background of chronic kidney disease [1] and in patients with end stage renal disease [3]. A subset of pRCC occurs in the setting of hereditary pRCC with a germline mutation of MET [4], but few sporadic cases also show MET mutations. Importantly, pRCC refers to a specific tumor entity, while “papillary growth/architecture” can be found in several renal tumors of different classes (see below for discussion).
pRCC were already reported in the first classification attempts by Ewing 1919, Melicow 1944 and Fite 1945 (for review see [5]), but terminology and diagnosis of this tumor type with his characteristic histological growth pattern has been modified over the last decades. In many classification proposals, different renal neoplasias were grouped together, regardless of histological architecture (papillary) or cytoplasmic staining (clear tumor cells). Manzilla Jimenez et al. described specific clinical and morphological features of papillary RCC and compared them with clear cell RCC in 1976 [6]. Thoenes et al. proposed in 1986 the Mainz renal tumor classification, based on cytoplasmic staining features [7]. They separated “chromophilic” RCC with mainly papillary architecture from clear cell and chromophobe RCC and proposed that “chromophilic” RCC originate from proximal tubule. In their classification, a basophilic small cell type and an eosinophilic (granular) cell type of “chromophilic” RCC were distinguished. Cytogenetic analysis of such tumors performed by Kovacs et al. showed characteristic trisomy and tetrasomy of chromosomes 7 and 17, and loss of the Y chromosome [8]. The validation of different renal tumor categories in the Mainz classification by these cytogenetic studies led to attempts for implementation of molecular findings in renal tumor classification.
Later, the Heidelberg classification [9] and Rochester Consensus Conference in 1997 formed the basis for the 2004 World Health Organization (WHO) classification with pRCC as an entity. Delahunt and Eble described in 1997 two major patterns of pRCC: one composed of small cells with scant basophilic to amphophilic cytoplasm and thin papillae lined by one row of tumor cells with inconspicuous nucleoli, frequently showing aggregates of foamy histiocytes and scattered psammomatous calcifications (type 1 pRCC), and another composed of cells with densely eosinophilic cytoplasm, larger nuclei with prominent nucleoli and more complex papillae with stratification of tumor cells (type 2 pRCC) [10, 11]. This was supported by molecular studies, describing that type 1 pRCC showed more consistent and uniform gains in chromosome 7 (encoding the MET gene) and 17 and loss in the Y chromosome, while type 2 pRCC were more heterogeneous and associated with additional gains in chromosomes 12, 16 and 20 [12]. Type 1 pRCC had a significant better prognosis than type 2 pRCC [12]. Different clusters of molecular signatures were also demonstrated which seemed to confirm that type 1 and 2 pRCC have a different molecular background [13–16]. Therefore, this taxonomy was introduced in the 2004 WHO renal tumor classification [17] and confirmed in the 2016 WHO renal tumor classification [18].
In the following years, various new renal tumor entities with papillary growth were introduced and tumors with an unequivocal genotype-phenotype correlation were better described, e.g. fumarate hydratase-deficient RCC, mucinous tubular and spindle cell RCC (MTSCC), tubulocystic RCC and TFE3- and TFEB-rearranged RCC. Clear cell papillary RCC (ccpRCC), also known in the literature as clear cell tubulo-papillary RCC, was also introduced, but is presently considered an indolent tumor in the current 2022 Classification (see below the section on “papillary tumors with clear tumor cells” for details). Over the years, it became clear that morphological type 1 and type 2 subtyping was controversial because in well-sampled tumors often mixed patterns exist impeding such subclassification and generating substantial interobserver variability [19–21]. Further, higher WHO/International Society of Urological Pathology (ISUP) grade in type 2 pRCC, and consequent association with histological aggressive features [22, 23] was not translated into worse patient outcome in multivariable analysis, and hence the prognostic value of such subtyping of pRCC was of no independent clinical value [24–26], which raised the hypothesis that type 1 and 2 may actually simply represent a WHO/ISUP grade progression phenomenon [20]. Most importantly, it was shown that type 2 pRCC consisted of at least three individual subgroups on the basis of molecular differences associated with patient survival [27]. Therefore, the 2022 WHO Classification no longer recommends subtyping pRCC into types 1 or 2 [28].
DIFFERENTIAL DIAGNOSIS OF RENAL TUMORS WITH PAPILLARY FEATURES
Papillary features/architecture can be observed in almost all renal tumor entities, even outside the spectrum of RCC [29] (including, for example, urothelial tumors of the kidney, metanephric tumors, mixed epithelial and stromal tumors, juxtaglomerular cell tumors, among others). Also, not all pRCC show an overt papillary architecture, which can be rather tubulopapillary or even hardly apparent and replaced by solid growth [29, 30]. In other words, there is a marked difference between having a papillary architecture / growth pattern, which can be observed in multiple renal neoplasms, and between calling a renal tumor pRCC (which refers to a specific tumor entity with defined criteria, and showing gains in 7/17 and loss of Y chromosomes). Papillary adenomas are currently defined upon the basis of size (<1.5 cm) as well as morphology (WHO/ISUP grade 1-2, unencapsulated) [1].
While there has been a lot of effort to subtype pRCC and segregate new entities, several patterns of growth and morphologies have been reported to impact prognosis across the pRCC spectrum, including solid, micropapillary, hobnail and microcystic patterns [26, 31]. WHO/ISUP grading and architecture remain strong predictors of tumor behavior and prognosis, and should always be reported [32]. Molecular features of pRCC are somewhat heterogeneous, reflecting the evolution in classifications. Besides the aforementioned typical chromosomal gains in 7/17 and MET mutations (which can be targeted by MET inhibitors [33, 34]), loss of CDKN2A has also been associated with aggressive behavior [35]. Aberrations in genes involved in mTOR and p53 pathways (mTOR, PIK3CA, PTEN or FBXW7, RB1, and TP53) support the involvement of these in the genesis and progression of pRCC [36, 37].
The differential diagnosis of pRCC encompasses tumors with basophilic cells, with clear cells, with eosinophilic cells and tumors with a mixture of eosinophilic and clear cells.
Papillary tumors with basophilic tumor cells: One of the entities that has stepped off the spectrum of the former type 1 pRCC is MTSCC. MTSCC are characterized by a compact tubulopapillary or cord-like growth pattern and low-grade spindle and anastomosing cells. Importantly, immunohistochemistry may not aid in the differential since it substantially overlaps (with AMACR and CK7 positivity in both entities), although CD10 is reported as being negative or only focal in MTSCC, while it is often multifocal/diffuse in pRCC [38]. Recently, VSTM2A expression by in situ hybridization has been identified as a biomarker of MTSCC, but it is not widely available [39]. To date, the main reason for separating these two entities is the different cytogenetic background (as MTSCC lacks the gains of chromosome 7/17 or loss of Y typical of pRCC), and different pathway activation (specifically with dysregulation of the Hippo pathway in MTSCC) [40–42]. In particular, MTSCC exhibit multiple chromosomal losses including chromosomes 1, 4, 6, 8, 9, 13, 14, 15 and 22, while pRCC with overlapping features predominantly showed multiple chromosomal gains [40]. Hence, molecular and/or copy number variation studies (including karyotyping) may be relevant in challenging cases.
In the event of a very solid pattern of pRCC with basophilic cytoplasm, the differential with metanephric adenoma (or epithelial-predominant nephroblastoma) is also challenging. Metanephric adenoma is composed of very compact tubules, branching ducts and abortive glomeruli, often with psammomatous calcifications, which mimic a tubulopapillary growth of pRCC [43]. Clues for the diagnosis include the higher nuclear-to-cytoplasmic ratio, a thick scarred-like/hyalinized stroma and absence of a fibrous capsule in metanephric adenoma [44]. The differential is solved by immunohistochemistry, as metanephric adenoma shows diffuse WT1 and CD57 positivity, negative or focal CK7 staining and usually positive BRAF staining [45], given the frequent BRAF V600E mutations [46]. Nephroblastoma is usually a triphasic tumor (although not infrequently biphasic or even monophasic), with higher mitotic index, being CD57 negative and usually showing no BRAF staining [43]. The clinical context, including age, associated syndromes, and presence and pattern of metastases is important for the diagnosis [47]. However, a subset of nephroblastomas do harbor BRAF V600E mutations and seem to arise from metanephric adenomas [48], so BRAF positivity does not completely rule out nephroblastoma.
Papillary tumors with clear tumor cells: The main differential diagnosis in this category is ccRCC, since ccRCC may have papillary or pseudopapillary foci admixed with the more classic growth patterns (solid, alveolar, acinar or microcystic). Importantly, and although most ccRCC are CK7 negative for only very focal, CK7 extensive staining may be found in ccRCC, especially in low-grade and cystic patterns [49]. In this context, there is also the differential diagnosis with clear cell papillary renal cell tumor (ccpRCT, formerly called “carcinoma”). In the 2022 WHO classification, this neoplasm is called “tumor” given the overall good outcome with no metastatic events in strictly diagnosed cases [50]. ccpRCT shows a tubule-papillary or cystic pattern (raising the differential with multilocular cystic neoplasm of low malignant potential) [51]. The tubulo-papillary pattern may also raise the differential with tumors showing leiomyomatous stroma and occurring in the setting of TSC and ELOC mutations [52, 53]. GATA3 is a helpful marker, being positive in ccpRCC, but it is not 100% sensitive nor specific [54]. Additionally, CK7 is diffuse, CD10 is negative, and CAIX shows a cup-shaped distribution which is helpful for diagnosis [55]. AMACR is also of use, being negative in ccpRCT and almost always diffusely positive in pRCC [56].
Papillary tumors with eosinophilic tumor cells: An entity that was for many years part of the former type 2 pRCC spectrum is FH deficient RCC (now designated as such to acknowledge that some tumors show somatic inactivation of FH but are not present in the germline). Identifying this phenotype is important because of the possibility of diagnosing the hereditary leiomyomatosis and renal cell cancer syndrome (HLRCC). Genetic counselling is advised to all patients with such tumors. These tumors are aggressive malignancies, often presenting with extra-renal extension and metastatic dissemination [57]. Tumor cells have high-grade nuclei (although low-grade forms have been reported [58]), with prominent cherry-red nucleoli surrounded by a clear halo. A low threshold for performing staining for FH is indicated [59]. Molecular testing for FH mutations is warranted in challenging cases [60].
Tubulocystic RCC is a rare tumor with a typical spongy gross appearance. Tumor cells show prominent hobnailing [61]. Tubulocystic RCC is usually an indolent tumor with a good prognosis. ALK-rearranged RCC is very rare and usually a diagnosis of exclusion. This tumor may include also papillary features [62]. Metastatic ALK-rearranged RCC have had dramatic responses to targeted ALK inhibitors. Collecting duct carcinoma and SMARCB-deficient medullary carcinoma have poor prognosis and often show a tubulopapillary growth, possibly causing problems in the differential diagnosis with pRCC. Features in favor of collecting duct carcinoma include a high-grade cytology and severe desmoplasia, with tubules and papillae showing an infiltrative growth pattern [63]. Admixture with more solid and trabecular growth is commonly found.
Papillary tumors with clear and eosinophilic tumor cells: Whenever there is an admixture of clear and eosinophilic cells, a tumor such as TFE3-rearranged RCC or TFEB-altered RCC should be considered [64, 65]. The definitive diagnosis still relies on break-apart fluorescence in situ hybridization (FISH) or next generation sequencing (NGS) [66]. Most TFE3-rearranged RCCs show a mixture of patterns, with solid, alveolar and papillary areas. Frequently one can find several psammomatous calcifications [64]. While it represents around 50% of pediatric RCCs, it is not infrequently found later in life [67], especially in young women [68]. Additionally, they can express (often focally) melanocytic markers such as Melan A or HMB45 [69]. TFEB-rearranged RCC are much less common than TFE3-rearranged RCC, they are much more frequently restricted to younger patients and they show a more indolent clinical behavior [70, 71]) [72]. Expression of melanocytic markers is more common in these, compared to TFE3-rearranged RCC [73]. Finally, TFEB-amplified RCC was also rarely reported. Importantly, they are aggressive tumors, with poor patient outcome [74, 75]. Interestingly, TFEB expression has been shown to correlate with PD-L1 expression in in vitro and tissue studies, and TFEB was suggested to mediate resistance to mTOR inhibitors by potentiating immune evasion, giving rationale to targeting both PD-L1 and mTOR simultaneously in the treatment of these tumors [76–78]. For TFEB-rearranged or TFEB-amplified RCC, TFEB FISH can establish the diagnosis [20, 79].
PROVISIONAL/EMERGING TUMOR ENTITIES WITH PAPILLARY GROWTH
There is an emerging body of evidence, that is compelling but not sufficient for placing certain clinicopathological and genomically distinct tumors into the formal classification. The 2022 WHO Classification currently regarded some papillary neoplasms as emerging entities [80]. One is biphasic squamoid-alveolar RCC, composed of one population of larger eosinophilic cells with higher nuclear grade arranged in nests, encased by a second population of smaller cells with high nuclear-to-cytoplasmic ratio, lower nuclear grade and basophilic cytoplasm, giving a low-power appearance of alveoli [81]. A quite characteristic features is the finding of emperipolesis, with engulfment of inflammatory cells by the larger cells [82]. Staining for cyclinD1 and CD57 specifically highlights the larger cells [83, 84]. Cytogenetically, however, tumors show the typical gains in chromosomes 7/17 found in pRCC [82, 85], as well as a high prevalence of MET alterations [86], supporting a link towards pRCC. The second subtype is the biphasic hyalinizing psammomatous RCC. These tumors are composed of one population of very small cells with hyperchromatic nuclei and somewhat spindle-shaped arranged around basement membrane material admixed with a second population of larger cells forming tubules, papillae or acini [87]. EMA stains preferentially the smaller cells, while CK7 highlights the larger cells. Interestingly, these tumors have been shown to harbor mutations (with biallelic loss) in the NF2 gene, and loss of the gene product Merlin [88], arguing in favor of an independent entity.
Papillary neoplasm with reverse polarity (PRNRP) has been increasingly reported in the literature [89, 90]. These are overall small cystic tumors, composed of thin papillae with edematous or hyalinized cores covered by a single layer of eosinophilic small cells with small low-grade nuclei (sometimes with optical clearing) pushed against the apical membrane (“reverse polarity”). Like for ccpRCT, GATA3 nuclear staining is characteristic and no worrisome features or poor outcome are reported [90]. Interestingly, the tumors have been associated with KRAS mutations in a high percentage of cases [91, 92].
Thyroid-like follicular RCC, which shares remarkable morphologic resemblance with follicular cancers of the thyroid gland are composed of variably sized follicles lined by small cuboidal cells. Papillary foci have been described [93], but usually a rather follicular architecture suggests the diagnosis. While most cases are indolent, aggressive behavior has been documented. Recently, these tumors have been shown to have a characteristic EWSR1::PATZ1 fusion [94–96]. The second one is the so-called Warthin-like pRCC, a pattern composed of densely eosinophilic papillae with oncocytic features, disposed on a stroma with brisk lymphocytic infiltrate, resembling a Warthin tumor of the salivary glands [97].
Table 1 summarizes the most frequent differential diagnoses of pRCC in routine practice and some features helping assisting in diagnosis, and Fig. 1 illustrates some of the phenotypes that come in the differential of pRCC.
Differential diagnosis of papillary renal cell carcinoma.

Abbreviations: FISH – fluorescence in situ hybridization; IHC – immunohistochemistry; ISUP – International Society of Urological Pathology; NGS – next generation sequencing; WHO – World Health Organization.
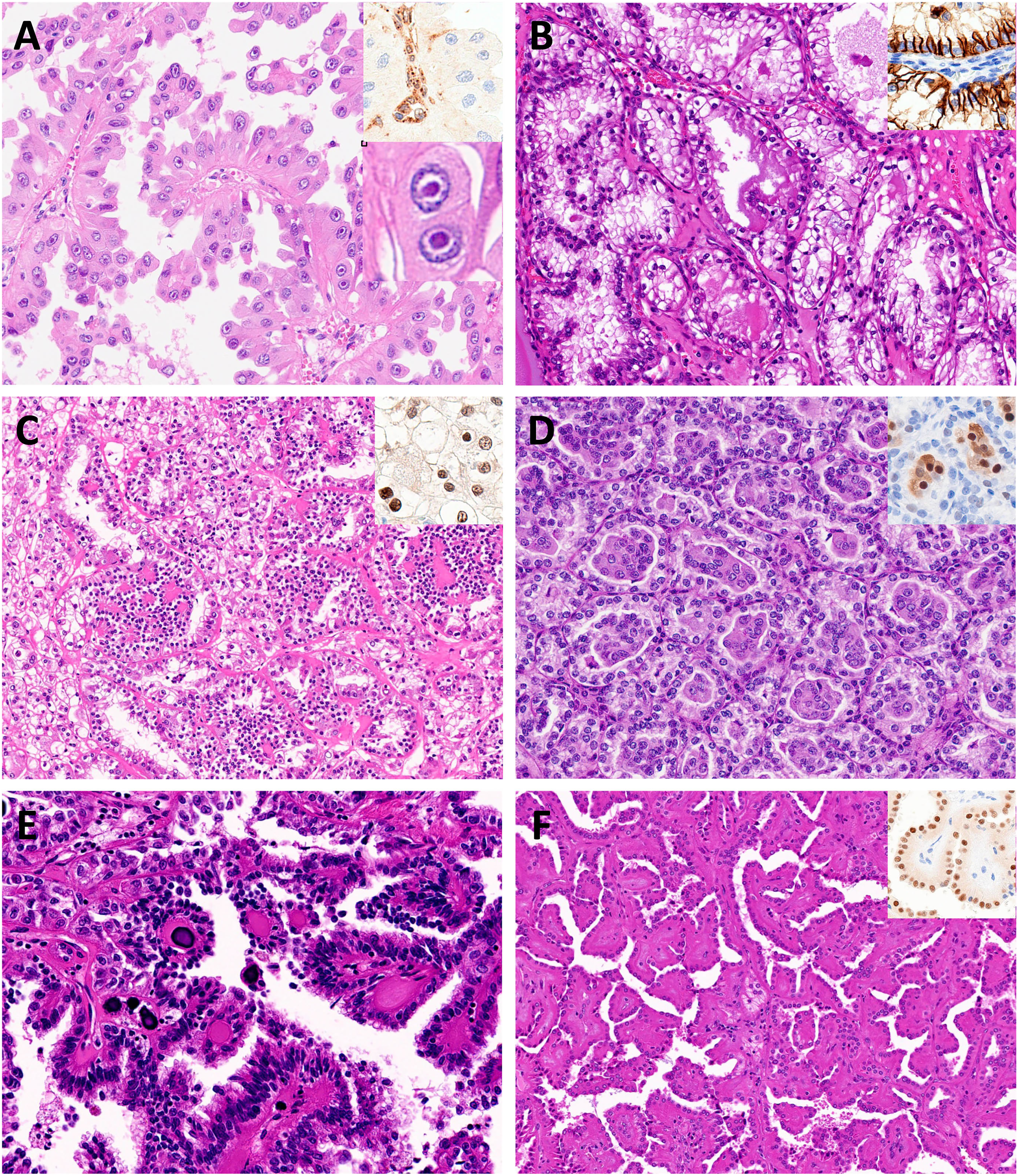
The examples of renal neoplasia with papillary architecture. A – FH-deficient RCC, showing papillary structures covered with cells with abundant eosinophilic cytoplasm and at least focal eosinophilic macronucleoli, often shows perinucleolar haloes (inset). B – Clear cell papillary renal cell tumor, showing tubulopapullary architecture lined by cuboidal cells with clear cytoplasm. The low-grade nuclei often exhibit linear arrangement apart from the basement membrane. The tumor cells show cup-like staining for CAIX with no luminal border staining (inset). C – TFEB-rearranged RCC, showing a biphasic pattern comprising nests of larger and smaller tumor cells surrounding basement membrane material. The tumors show diffuse TFEB nuclear positivity (inset). D – Biphasic squamoid alveolar RCC, showing a biphasic pattern with squamoid cells with abundant eosinophilic cytoplasm in the center of the alveolar structures. Squamoid cells are positive for cyclinD1 (inset). E – Biphasic hyalinizing psammomatous RCC, showing biphasic pattern with tubulopapillary growth of larger cells and also showing smaller cells, with psammomatous calcifications and having a glomeruloid pattern. F – Papillary renal neoplasm with reverse polarity with thinly branching papillae lined by eosinophilic cells with low-grade nuclei aligned at the apex. The tumors show diffuse GATA3 nuclear positivity (inset).
FUTURE PERSPECTIVES
In conclusion, the spectrum of pRCC has changed in recent years, with many entities being identified. The 2022 WHO renal tumor classification has introduced the concept of molecularly defined RCC entities and abolished the separation of type 1 and type 2 pRCC. Several new renal tumor entities with papillary growth are emerging. Therefore, it is expected that the spectrum of pRCC is becoming increasingly narrow (Fig. 2). Continuing revisiting tumors with papillary features and gathering data on particular patterns, combined with accurate genotype-phenotype and clinical correlation, will contribute to precision medicine and better patient risk stratification and selection for specific therapies [98].

The spectrum of papillary renal cell carcinoma is narrowing. Comparison of the WHO 2016 and WHO 2022 classification of papillary renal cell carcinoma.
Although several targeted therapy options are possible for patients with metastatic pRCC, many new therapeutical options have been approved based on trials including all histotypes of RCC (mainly ccRCC). Data specifically regarding pRCC is scarce [99]. In this sense, accurate histopathological characterization of renal cell tumors is important to identify specific phenotypes which may have a distinct and actionable molecular background. With the introduction of molecularly defined entities, pathologists should attempt to accompany this evolving field and acquire tools, such as dedicated NGS panels, for characterizing challenging and heterogeneous renal cancers [100]. Also, application of digital pathology algorithms for dissecting architectural and cytological features of renal cancers with papillary foci will also likely contribute to uncovering novel biomarkers to integrate with molecular data, and may be a useful tool to deconvolute the heterogeneity of renal tumors, facilitating diagnosis and predictive value of specific biomarkers [101–103].
Footnotes
ACKNOWLEDGMENTS
The authors have no acknowledgments.
AUTHOR CONTRIBUTIONS
Drafting of the paper, table and figure: João Lobo and Riuko Ohashi.
Supervision and final editing: Holger Moch.
All authors read and approved the final paper.
FUNDING
H.M. receives a Swiss National Science Foundation grant (No. S-87701-03-01) and R.O. receives a JSPS KAKENHI Grant Number JP22KK0273 (Fostering Joint International Research (A)).
CONFLICTS OF INTEREST
J.L., R.O. and HM. have no conflicts of interest to disclose.