
Abstract
RCC has witnessed a significant increase in its incidence over the last five decades, ranking as the ninth most common cancer globally. Although survival rates have improved substantially, RCC remains one of the deadliest urological cancers. Traditionally, RCC subtypes were classified based on histopathological features. However, in recent years, there has been a paradigm shift towards molecular and genomic characterization of RCC, leading to the recognition of distinct molecular subtypes.
The 2022 World Health Organization (WHO) classification introduced a new category called “molecularly defined renal carcinomas,” encompassing various subtypes, including SMARCB1-deficient medullary carcinoma, ALK-rearranged RCC, FH-deficient RCC, SDH-deficient RCC, ELOC-mutated RCC, TFEB-altered RCC, and TFE3-rearranged RCC.
These molecular subgroups have significant consequences for diagnosis, prognosis, and treatment. Molecularly defined RCCs are frequently underrepresented in clinical trials, encouraging additional research to identify beneficial therapeutics. Immune checkpoint inhibitors and tyrosine- kinase inhibitors have shown promising results in some subtypes, while others may benefit from specific inhibitors targeting their molecular drivers. Additionally, these classifications have important prognostic implications, guiding treatment decisions and genetic counseling.
Keywords
INTRODUCTION
Renal cell carcinoma (RCC) has doubled its incidence in the last fifty years becoming the ninth most frequent tumor in the world (2% of global cancer diagnoses). Even though, its survival rate has considerably improved in the last half century (76% 5-year survival rate in the US for years 2009–2015 vs. 46.8% in 1977) it is considered as one of the deadliest urological cancers [1].
Traditionally, renal tumor subtypes have been classified based on histopathological characteristics. The majority of RCCs present as clear cell (ccRCC), papillary (pRCC), and chromophobe (chRCC) histological subtypes. Among these, ccRCC stands out as the most prevalent and clinically aggressive variant [1].
However, in recent years, there has been a shift towards emphasizing molecular and genomic features. The Cancer Genome Atlas (TCGA) published in 2013 included the molecular peculiarities of RCC [2]. This has been driven by our deeper understanding of the molecular landscape of RCC and the recognition of the clinical significance of specific molecular alterations.
As a result, the latest edition (5th edition) of the “World Health Organization (WHO) classification of urinary and male genital tumors”, released in 2022, introduced a new category known as “molecularly defined renal carcinomas”. Such molecularly defined epithelial renal tumors include SMARCB1 (SWI/SNF-related matrix-associated actin-dependent regulator of chromatin subfamily B member 1)-deficient medullary RCC, Anaplastic lymphoma kinase(Alk)-rearranged RCC, Succinate dehydrogenase (SDH)- deficient RCC, Fumarate hydratase (FH)-deficient RCC, Elongin C (ELOC)-mutated RCC, TFEB (Transcription factor EB)-altered RCC and TFE3 (Transcription factor E3)-rearranged RCC [3]. These last two entities (TFEB and TFE3 altered RCCs) had been formerly described in literature as translocation RCC (tRCC) or MiT/TFE Family RCC.
This implies a transition from a morphology-based classification system to a molecularly oriented classification. However, for a precise diagnosis of RCC, it is essential to consider not only molecular features but also radiological, clinical, and morphological characteristics, assessed through light microscopy (LM) and immunohistochemistry (IHC) techniques [3].
We are thus confronted with a subset of entities that have limited representation in medical literature and clinical trials, resulting in a general lack of experience. Therefore, we will now outline the key clinical features for a better understanding of these new entities.
CLINICAL, MOLECULAR AND HISTOPHATOLOGICAL CHARACTERISTHICS
SMARCB1-deficient medullary carcinoma
In the latest edition of the WHO classification, renal medullary carcinoma has been renamed as SMARCB1-deficient medullary carcinoma. This change in nomenclature reflects the hallmark features of these tumors: the complete loss of nuclear expression of SMARCB1 (INI1 SNF5, BAF47) [3].
SMARCB1 plays a crucial role in various cellular processes, including DNA damage repair, DNA replication, proliferation, and differentiation. The driving molecular event underlying SMARCB1 protein deficiency is commonly a combined translocation and hemizygous gene loss, although homozygous gene deletion and, more rarely, pathogenic somatic mutations can explain the lack of expression of SMARCB1 in other cases [4].
The diagnosis of renal medullary carcinoma, now termed SMARCB1-deficient medullary carcinoma, is typically established by detecting the absence of SMARCB1 (INI1) expression in tumor cells through IHC.
These tumors are highly aggressive, with a median overall survival of 13 months [5]. SMARCB1-deficient medullary carcinomas are typically symptomatic tumors that almost exclusively develop in the renal medulla of young patients of African descent with hemoglobinopathies, such as sickle cell trait. However, SMARCB1 protein loss has been described in other, non-hemoglobinopathy associated, cases of rare renal carcinomas with medullary phenotype, highlighting the underlying molecular alteration as a common defining characteristic of the category [4, 6].
ALK-rearranged RCC
ALK- rearranged RCC occur at frequencies of less than 1% of RCC. This entity was first described in 2011 by Debelenko et al. [7]. ALK-rearranged RCC are characterized by the rearrangement of the anaplastic lymphoma kinase (ALK) gene (located on the 2p23 chromosome) with a fusion partner such as VCL, TPM3, EML4, HOOK1 and STRN gene [7].
Among these plausible fusion partners, literature splits ALK-rearranged RCCs into two distinct categories based on whether VCL works as a fusion partner or not. These differentiation obeys to different clinical behaviors [8].
VCL-ALK-RCCs are typically observed in younger patients, although they can occur in individuals of other age groups as well. They are notably associated with sickle cell trait. These tumors manifest unique histological characteristics, including polygonal cells featuring vesicular nuclei and an abundance of cytoplasm marked by prominent cytoplasmic vacuoles [8].
In contrast, non-VCL-ALK RCCs exhibit a wider age distribution and present a more diverse morphological spectrum. This includes papillary, solid, tubular, tubule-papillary, cribriform, signet ring cell, metanephric adenoma-like, and mucinous tubular and spindle cell-like patterns [8].
There are multiple mechanisms that can cause ALK hyperactivation such as gene amplification, translocations involving the kinase domain of the protein and point mutations [9].
ALK rearrangements can be identified using molecular techniquesincluding fluorescence in situ hybridization (FISH) or next-generation sequencing (NGS) [7].
Targeted therapy, such as ALK inhibitors, have transformed the therapeutic landscape for ALK-rearranged RCC. These drugs specifically target the abnormal ALK fusion proteins, disrupting their signaling pathways and inhibiting tumor growth. Hence, precise diagnosis of ALK-rearranged RCC are crucial in order to determine suitable treatment options and optimize patient outcomes [10–13].
FH-deficient RCC
In the 2016 edition of the WHO classification, hereditary leiomyomatosis and RCC (HLRCC) syndrome-associated RCC was recognized as a distinct entity [14]. HLRCC is characterized by a predisposition to aggressive RCC, as well as the presence of uterine and cutaneous leiomyomas. The autosomal dominant germline mutations in the fumarate hydratase (FH) gene serve as the fundamental cause of HLRCC.
Nevertheless, several studies have revealed the existence of somatic modifications in the FH gene, leading to the deficiency of FH protein, even in individuals without a documented history of HLRCC. This posed challenges in terms of classifying this group of tumors. As a result, the WHO 2022 classification introduced the term “FH-deficient RCC” to encompass this subgroup [3, 15].
FH-deficient RCC often displays heterogeneous and mixed pathological architectural patterns [16]. Diagnosis of this entity can be established with a negative FH IHC staining, which is highly specific, in conjunction with positive nuclear and cytoplasmic IHC staining for S-(2-succino)-cysteine (2SC) [16, 17]. Ultimately, genetic germline testing for a pathogenic FH mutation would determine the association with a HLRCC syndrome.
Intracellularly, the FH deficiency results in a Krebs cycle and oxidative phosphorylation impairment in these tumors. Consequently, there is an increase of intracellular fumarate and oxidative stress resulting in an accumulation of the hypoxia-inducible factor (HIF). This, finally, leads to an increased vascular endothelial growth factor (VEGF) activity. At the same time, aerobic glycolysis is promoted through activated epidermal growth factor receptor (EGFR) signaling [18]. Increasing knowledge regarding metabolic alterations in this particular entity has led to developing promising therapies targeting these two forementioned pathways.
Clinically FH-RCCs are highly aggressive, and most cases present with locally advanced or metastatic disease. In general, patients experience an unfavorable prognosis. A retrospective case series with 32 patients recently showed that after a median follow-up of 16 months only 50% of patients were alive [16].
SDH-deficient RCC
SDH-deficient RCC is rarely seen without a SDH germline mutation, being the most prevalent SDHB germline mutation. SDHC, SDHA or SDHD mutations are less frequent. Germline mutations in SDH subunit genes are often associated with various other tumors and conditions, including paraganglioma, pheochromocytoma, type 2 gastrointestinal stromal tumor (GIST), and pituitary adenoma [18].
SDH-deficient RCC exhibits distinct morphological features under a microscope. These tumors characteristically show cytoplasmic vacuoles or inclusions, representing bigger mitochondria. Cytoplasm is usually eosinophilic [19]. SDH-deficient RCC often show low grade cytological characteristics. These cases are rarely metastatic (12%), being surgical removal curative in most cases, while in high grade SDH-deficient RCC disseminated disease is seen in up to 70% of patients [18, 20].
Diagnosis of SDH-deficient RCC is typically confirmed with IHC testing. Loss of expression of one of the SDH subunits in tumor cells is a key diagnostic feature [21].
The SDH enzyme complex plays a crucial role in linking the Krebs cycle and the electron transport chain in the mitochondria. Similarly to FH-deficient tumors, SDH dysfunction leads to the stabilization of HIFs. This can promote tumorigenesis through a pseudohypoxic pathway, comparable to the way (the) VHL gene dysfunction contributes to kidney cancer.
Because of the underlying molecular mechanisms involving SDH dysfunction and HIF stabilization, tyrosine kinase inhibitors (TKIs) may be considered as a treatment option for SDH-deficient RCC [18, 21].
ELOC mutated RCC
ELOC (formerly known as TCEB1)-mutated RCC has emerged as a new entity within the WHO 2022 molecularly defined RCCs. These tumors show a broad morphologic spectrum, most presenting thick, peripheral fibromuscular bands with clear cell intercalated areas. Diffuse positive CK7 staining is usually observed. These characteristics significantly overlap with those of sporadic tumors harboring TSC1 (tuberous sclerosis 1)/TSC2 and MTOR mutations, as well as identical tumors occurring in patients with TSC syndrome [22].
Before The WHO 2022 renal tumor classification, the GUPS (Genitourinary Pathology Society) consensus recognized RCC with fibromyomatous stroma according to its morphologic and IHC similarities, despite their different molecular hallmarks [23].
Similarly, RCC with leiomyomatous stroma (RCCLMS) was included as a provisional entity in the 2016 World Health Organization (WHO) classification of renal carcinomas [14].
Notably, mutation on ELOC gene is now considered an essential diagnosis criterion for ELOC-mutated RCC, making molecular characterization mandatory to identify these rare tumors [6, 18].
Typically, ELOC-mutated RCCs exhibit an indolent clinical course after tumor resection. However, due to the limited number of fully characterized cases, caution is warranted regarding the prognostic implications associated with these tumors [18, 24]. TSC/MTOR mutated RCC show similar, indolent clinical courses although further studies should be addressed before drawing prognostic considerations [22].
TFEB-altered and TFE3-rearranged RCC
Formerly considered a single entity, known as translocated RCC (tRCC) or MiT/TFE Family RCC, different morphological, molecular and clinical characteristics has led to consider TFEB-altered RCC and TFE3-rearranged RCC as two independent malignancies [3, 25].
TFE3-rearranged RCC
TFE3 rearrangement, being Xp11 translocation the most described gene alteration, is one of the most common molecular driver alterations seen in RCC [23]. These tumors are characterized by fusion events involving the TFE3 gene located on the Xp11 chromosome and partner genes found on chromosomes 1, 17, and X. Some frequently observed fusion partners include ASPSCR1, PRCC, and SFPQ. TFE3 fusion genes may enhance signaling pathways that as a result promote tumorigenesis, such as the mTORC1 pathway upregulation and increase of TGF-beta levels. There is also a dysregulation of CD40L helping tumor immune escape [26–29].
TFE3-rearranged RCC are more commonly seen in the pediatric population, accounting for approximately 40% of all diagnosed renal cancer cases in children, and typically exhibit an indolent course. In contrast, these tumors are less frequently observed in adults, representing only 1–4% of cases. However, given the higher overall incidence of renal cancer in adults compared to children, TFE3-rearranged RCC are in absolute numbers more common among adults. In adult cases, TFE3-rearranged RCC are considered highly aggressive, particularly among younger adults [3, 29].
While Fluorescent in Situ Hybridization (FISH) is considered the gold standard test for diagnosing this condition (excluding some intrachromosomal rearrangements: RBM10-TFE3 gene fusion), the typical diagnostic approach involves the use of IHC tests [29, 30]. However, recent studies underscore that IHC may not serve as an effective surrogate for molecular techniques due to its lack of specificity. The C-terminus binding site, targeted by the TFEB antibody, has been proven not to be as sensitive and specific for TFE3-rearranged neoplasms as previously believed [31]. For instance, authors have described several cases of renal cell carcinoma with positive TFE3 expression by IHC that did not exhibit the same positivity when assessed using molecular techniques, strongly suggesting that TFE3 immunohistochemistry lacks value in distinguishing this condition [32–34].
RNA sequencing and Next-Generation Sequencing (NGS) can also be used to identify the fusion transcript and the genes involved in TFE3-positive RCC. Nevertheless, not all molecular panels include a TFE3 analysis and this should be considered when NGS panels Fig. 1.
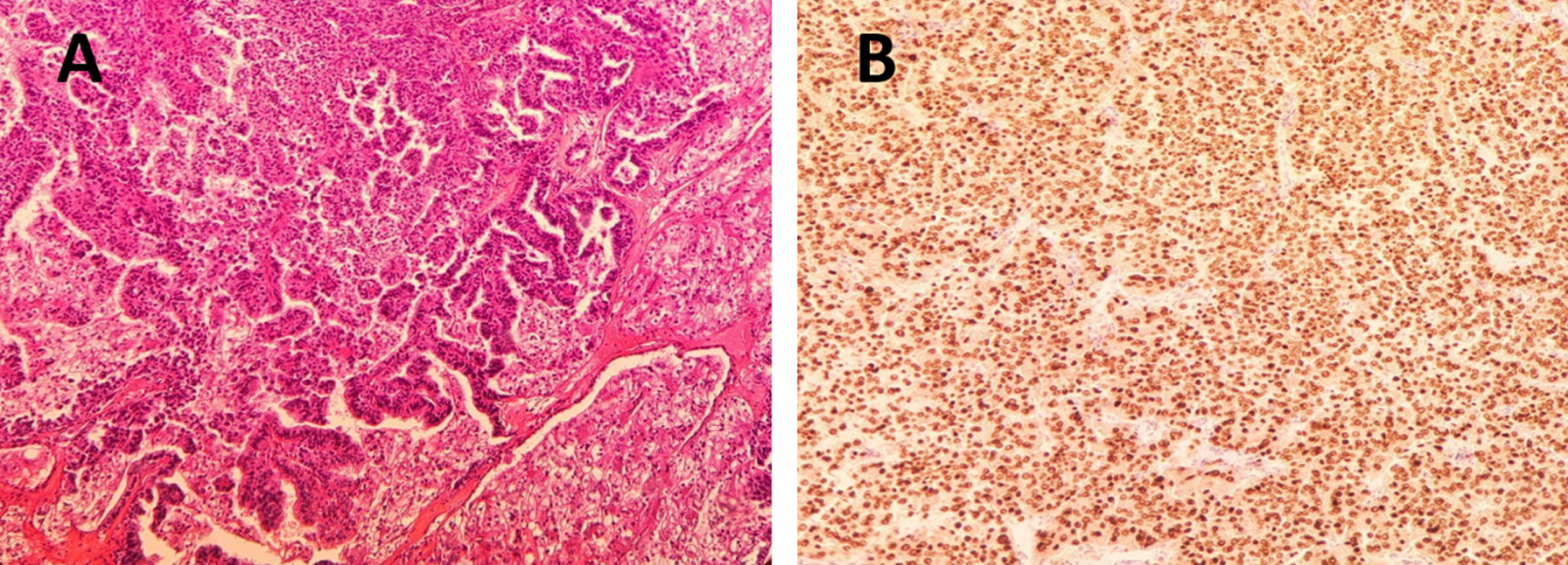
Unfortunately, as of the present date, there are no specific treatment recommendations available. Immune checkpoints appear to yield better results compared to antiangiogenics, as evidenced by the findings from the IMmotion 151 study [35]. Cabozantinib seems to be the exception, probably due to its direct effect of inhibiting MET which is among the few oncogenic drivers of TFE3- rearranged RCC [36–38].
TFEB-altered RCC
TFEB-altered RCC is less prevalent than TFE3 RCC. It involves at [6, 11] (p21; q12) translocation or amplification resulting in TFEB-rearranged or TFEB-amplified RCC, respectively. Each exhibits distinct characteristics. It’s noteworthy that certain RCC cases may involve a TFEB rearrangement within the context of a 6p21.1 amplification [39].
Clinically, TFEB-rearranged RCCs are usually indolent, with low-stage tumors observed in young patients (median age 31 years) [39]. In contrast, TFEB-altered RCCs manifest at an older age (median age 64.5 years) [39]. They are considered aggressive tumors, often presenting with locally advanced disease and an elevated likelihood of developing distant metastases [23, 40].
Morphologically, TFEB-rearranged RCCs are classically described as biphasic, with both large and small epithelioid cells grouped around eosinophilic spheres composed of basement membrane material. However, various other heterogeneous morphologies have been identified, such as nuclear pseudoinclusions, extensive hyalinization, and psammomatous calcifications [39, 40]. Alternatively, TFEB-amplified RCCs rarely exhibit a biphasic morphology, displaying greater variability while consistently maintaining a high nucleolar grade [39, 40].
There is limited information regarding the treatment of this tumor subtype. Nevertheless, TFEB-amplified RCCs are associated with VEGFA coamplification due to their proximity in the gene region. In this context, Gupta et al. evaluated VEGF-targeted therapy (n = 4), with one patient experiencing a prolonged response to treatment. Despite the limited number of patients, not sufficient to establish its effectiveness, it suggests a potential therapy in this aggressive setting [41, 42].
Furthermore, in a preclinical setting, Zhang et al. demonstrated that TFEB mediates immune evasion and resistance to mTOR inhibition via the induction of PD-L1 expression. Thus, immune checkpoint inhibitors (ICI) and mTOR-targeted therapy appear to be suitable treatment options for this population [43].
Being part of the same cluster, they appear as two completely different entities, prompting consideration of whether they should be differentiated in clinical trials. Come what may, the development of clinical trials in this scenario is vital to find the better therapeutic approach.
THERAPEUTIC IMPLICATIONS IN 2022 WHO MOLECULARLY DEFINED RCC
Most of these newly described entities have been recently differentiated from other RCC subtypes. This, added to the fact that the majority of molecularly defined renal cell carcinomas are considered rare, makes them broadly underrepresented or even undetected in clinical trials. Most evidence concerning these RCC subtypes has been collected retrospectively in RCC clinical trials populations, giving us a glimpse of the behavior of molecularly defined RCCs treated with current RCC approved therapies (Table 1). However, their inclusion in WHO’s 2022 classification has brought interest to this group of entities and we can expect them to be more present in current and future clinical trials protocols and even see trials being held for a specific group of RCC, driven by a potential molecular actionability. An exception is ALK-rearranged RCC which represents the paradigm of an alteration-driven tumor against which targeted therapies development is far ahead from other molecularly defined RCC.
Ended prospective and retrospective studies including tRCC.
PFS (Progresion Free Survival), OS (Overall Survival), HR (Hazard Ratio), RCC (Renal Cell Carcinoma), ccRCC (clear-cell Renal Cell Carcinoma), nccRCC (non-clear-cell Renal Cell Carcinoma),pRCC(PapilaryRenall Cell Carcinoma), ICI (Inmune- Checkpoints- Inhibitor), HLRCC(Advanced Hereditary Leiomyomatosis and Renal Cell Cancer) *Specific data about the molecularly defined RCC.
In the past, both VEGF-targeted and mTOR-targeted monotherapies have been extensively researched and constituted the established standard of care for non-clear-cell renal carcinoma.
However, in 2016, after the publication of the phase 2 clinical trials ESPN, which enrolled 68 patients, of which only 7 had Xp11.2 translocation-associated RCC, and ASPEN (N = 108), with 8 unspecified translocated RCC, multi-kinase inhibitor sunitinib stood against classical MTOR- inhibitor everolimus as the preferred option for treating non clear cell renal cell carcinoma. Nevertheless, results were poor showing a 5.9% ORR and 6.1 months median PFS in the ESPN trial and a 5.18% ORR and 8.3 months median PFS in the ASPEN trial [44, 45].
In the tRCC scenario VEGF targeted therapies efficacy was also studied. Malouf et al carried out a French retrospective study with 21 tRCC patients that had received sunitinib, sorafenib or everolimus. Median PFS was 11 months with sunitinib, 6 months with sorafenib and 3 months with everolimus [46]. In the same way, Chouieri analyzed outcomes of 15 tRCC patients treated with sunitinib, sorafenib, bevazicumab or ramucirumab. Median PFS of the whole cohort was 7.1 months with a 14.3 months OS median [47].
There may be a molecular explanation for TKIs and mTOR targeted therapies’ poor activity in tRCC: transcription factor NRF2 (nuclear factor erythroid-derived-2-like 2 [NFE2L2]) plays an important antioxidative role [48], and high NRF2 activity has been correlated to resistance to several drugs, including sunitinib, axitinib, lenvatinib or temsirolimus [49]. Characteristically, tRCC show high NRF2 activity. These findings are in line with poor outcomes of tRCC patients treated with TKIs and mTOR targeted therapies.
While TKIs activity against tRCC may seem discreet, cabozantinib, a VEGF-TKI with anti-MET activity has shown promising results. In the 2021 ASCO Genitourinary Congress the results of a multicenter, retrospective, international cohort study of cabozantinib in 52tRCC patients were released demonstrating a median PFS and OS of 6.8 and 18.3 months respectively [36].
Along with TKIs, immunotherapy has revolutionized RCC therapy for good. Retrospective data from trials testing immunotherapy alone or in combination with antiangiogenic agents or TKIs suggest immune checkpoint inhibitors (ICIs) may be an effective therapy in tRCC. Preclinical findings support this idea, showing a high density of tumor infiltrating CD8 T-cells in these tumors. Moreover, the proportion of non-exhausted and ICIs responsive CD8 + PD1 + TIM3–LAG3–T cells in these tumors is similar to what has been described in ccRCC, an ICI responsive tumor [50]. This could explain tRCC described ICI responsiveness despite an apparent lack of other immunogenic biomarkers such as a typically low tumor mutational burden [51].
The phase III IMmotion 151 clinical trial showed better PFS without improvement in OS with the bevacizumab plus atezolizumab combination compared to sunitinib in ccRCC or sarcomatoid histology RCC patients [35]. Specifically, an analysis of 15 patients with TFEB/TFE3 translocations enrolled in this study (6 received sunitinib, 9 received atezolizumab-bevacizumab) showed a significantly greater benefit in PFS with the atezolizumab-bevacizumab combination (median PFS 3.5 months with sunitinib versus 15.8 months with atezolizumab-bevacizumab).
Boilève et al. carried an international, multicenter retrospective study with 24 metastatic former known MITF family tRCC patients that had received ICI (nivolumab, pembrolizumab and ipilimumab). Overall survival was 24 months reflecting a good therapy response. Nevertheless, tumor burden mutation rate was analyzed in four patients showing a mutation rate of 4–30 mutations/exome. Overall, median mutational load of these 4 tRCCs was lower than that of the ccRCC samples from the TCGA dataset (n = 424; p < 0.0001). Consistent with previous described preclinical findings [51].
Following this direction, McGregor et al enrolled in a phase II study patients with clear-cell RCC with at least 20% of sarcomatoid differentiation and various non-clear cellRCC (nccRCC) histologies including chromophobe, papillary, medullary, collecting duct, TFE3 translocation, and unclassified RCC with or without sarcomatoid differentiation. Its primary end point was to determine the ORR of atezolizumab plus bevacizumab. Overall, ORR was 33%. In patients with ncc-RCC ORR was 26%. On top of that, 20% of the five TFE3 translocated patients showed an objective response [52].
Other combinations of ICI+anti vEGF therapies have also been tested. The COSMIC-021 trial tested cabozantinib plus atezolizumab in a multicenter, open-label, phase Ib study. The study population included a cohort of 32 patients with nccRCC among which 3 had molecularly-defined RCC (one fumarate hydrase–deficient RCC, one MiT-family translocation RCC, one unspecified translocated RCC). In the nccRCC cohort ORR was 31% and disease control rate was 94% [53].
CA209-9KU is a phase 2 clinical trial that tested another TKI-immunotherapy combination with cabozantinib plus nivolumab. Cohort 1 included 40 patients with papillary, unclassified, FH-deficient, or translocation associated RCC. Five patients with FH-deficiency and two patients with translocation associated RCC were included. Median PFS was 12.5 months and ORR was 48%. Moreover, an objective response was obtained by all FH-deficient and half translocated RCC carcinoma [54].
Finally, the phase 3b/4 clinical trial CheckMate 920 (N = 52) of nivolumab plus ipilimumab in treatment naïve patients for advanced or metastatic RCC included 2 patients with translocation-associated RCC which had stable and progressive disease as best response [55]. Similar results were encountered in the clinical trial CheckMate 374 (N = 44) which assessed the clinical activity of nivolumab in monotherapy. This trial included 2 patients with translocation-associated RCC, who had progressive disease as the best response [56].
These new RCC molecular-based subclassification should be taken in account not only for their potential actionability but for its prognostic implications. For instance, this classification should be integrated into the consideration of adjuvant treatment, particularly in cases involving less-aggressive categories like ELOC-mutated RCC or TFEB-rearranged RCC. Moreover, the identification of SDH-deficient RCC and FH-deficient RCC advances genetic counseling for patients and their families while expanding the therapeutic options for these individuals [18].
TARGETED THERAPIES
While it may be enticing to molecularly define these tumors in pursuit of actionable insights, the fact remains that there are still limited targeted therapies available for specific molecularly defined RCC subtypes.
Crizotinb was the pioneer ALK-i tested in RCC due to its ability to inhibit the mitogen-activated protein kinase (MET) which participates in the resistance mechanisms to anti-vascular endothelial growth factor (VEGF) therapies. Schöffski et al. described the positive outcomes of one of the cohorts of the biomarker-driven, single-agent, non-randomized, open-label, two-stage phase II CREATE trial. Patients with advanced renal-cell carcinoma type 1 with MET mutations or amplification were grouped considering the existence or not of MET alterations. The median OS was 30.5 months, with an OS for the MET negative patients of 14.5 months [57]. In 2021, a randomized phase II trial that aim to determine if MET-targeted therapy (cabozantinib, crizotinib or savolitinib) could improve clinical outcomes when compare to usual therapy with VEGF-directed therapies (sunitinib). Lamentably, the more selective MET inhibitors crizotinib and savolitinib did not appear to have superior clinical activity versus sunitinib. On top of that, they were halted early in the study due to a pre-planned futility analysis [58].
Unfortunately, literature comprises limited data about ALK-i in the ALK-rearranged RCC setting. For instance, Pal et al. described in 2018 three cases of EML4-ALK fusion RCC who had progressed to several prior therapies, including multi-target tyrosine kinase inhibitors (TKI) and immune checkpoint inhibitors (ICI), that were treated with alectinib reaching a deep clinical and radiological response [59]. Similar results were obtained in the case reports described by Varchetta et al. and Zhou et al. In short, ALK-i have shown radiological responses and an improvement of the performance status [21, 61].
Entrectinib is an ALK, ROS1, TrkA, TrkB, and TrkC inhibitor which has also shown encouraging results in treating RCC ALK- rearranged patients. The RXDX-101 (NCT02097810) phase I and IIA basket clinical trial included a 22-year-old patient with an early relapse of a VCL-ALK translocation RCC. Radiological and clinical results were soon obtained and maintained for 19 months [13].
Moreover, in the era of precision medicine where tumor agnostic therapies have emerged as a revolutionary paradigm of cancer treatment, ALK-i are earning an important role in the ALK-positive tumor agnostic setting. There are at least two clinical trials going on in this scenery, the CREATE trial (NCT01524926) and the Alpha-T trial (NCT03456076). On the one hand, the CREATE trial is a phase II study that evaluates the efficacy and safety of crizotinib in predefined tumor types with specific alterations in ALK and/or MET [62]. On the other hand, the Alpha-T trial is a phase II trial that analyses the antitumor activity of alectinib in patient with ALK positive solid tumors other than lung cancer [63].
Hence, ALK-rearranged RCC multicenter-randomized-clinical trials must be designed to confirm the initial evidence that suggest that ALK-i might benefit this subgroup of patients.
Other targeted therapies are being tested among some other molecularly defined RCC.
Following preclinical data showing VEGF and EGFR genes upregulation in FH-deficient tumors, a phase 2 clinical trial with erlotinib+bevacizumab was carried on. Results were published in ASCO 2022 showing an overall ORR of 51% with a median PFS of 14.2 months [64].
In the SMARCB1 setting the ongoing clinical trials involving proteasome inhibitors (NCT03587662) and EZH2 inhibitor tazemetostat(NCT02601950) offer promising avenues for the treatment of SMARCB1-deficient renal medullary carcinoma (RMC) and other SMARCB1-negative tumors.
EZH2 inhibition has shown promising results in preclinical studies. By inhibiting EZH2, a catalytic subunit of the polycomb repressor complex 2 (PRC2), researchers have been able to trigger cell death in malignant rhabdoid tumors, including those marked by SMARCB1 deficiency. This success is attributed to the capacity of EZH2 to antagonize the SWI/SNF complex, a critical player in cancer progression [65, 66].
Ongoing clinical trial in molecularly defined RCC are described in Table 2.
Ongoing clinical trial in molecularly defined RCC.
SMARCB1 (SWI/SNF-related matrix-associated actin-dependent regulator of chromatin subfamily B member 1)-deficient medullary RCC, Anaplastic lymphoma kinase (Alk)-rearranged RCC, Succinate dehydrogenase (SDH)-deficient RCC, Fumarate hydratase (FH)-deficient RCC, TFE3 (Transcription factor E3)-rearranged RCC *ST: standard dose paclitaxel in 28-day cycle, standard dose everolimus in 28-day cycles, standard dose erlotinib in 28-day cycles, standard dose docetaxel in 21-day cycles, standard dose cabozantinib in 28-day cycles.
CONCLUSION
What once seemed like a distant utopia has now become an everyday reality for many of us. The widespread availability and advancement of Next Generation Sequencing tools have ushered in the era of precision medicine, seamlessly integrating it into our clinical practice. Consequently, it is not merely advisable but absolutely essential to distinguish and characterize RCC’s molecularly defined carcinomas, even though they represent a relatively rare subtype of this disease. To address this imperative need, Molecular Tumor Boards have emerged as invaluable forums for facilitating interdisciplinary discussions and reaching a consensus on therapeutic decisions.
Furthermore, it is especially important that we foster and support the ongoing development of clinical trials within this context. These trials hold the promise of unlocking tailored treatments and improving outcomes for patients. Embracing these molecular advancements and promoting research efforts is not just a choice but an ethical obligation in our quest to combat cancer effectively.
Footnotes
ACKNOWLEDGMENTS
The authors have no acknowledgments.
FUNDING
The authors report no funding.
AUTHORS CONTRIBUTIONS
MAA: performed literature research; wrote and edited manuscript; PBM: wrote and edited manuscript; NVC: oversaw, edited and critical revision of the manuscript; IMP: oversaw, edited and critical revision of the manuscript; MTS: provided the images; JP: oversaw, edited and critical revision of the manuscript.
CONFLICTS OF INTEREST
MAA has no relevant disclosure; PBM has no relevant disclosure; NVC has provided consulting for Janssen and Astellas; IMP has received has received travel funding from Merk; MTS has no relevant disclosure; JP is an Editorial Board member of this journal, but was not involved in the peer-review process of this manuscript nor had access to any information regarding its peer-review: oversaw, edited and critical revision of the manuscript. JP has also participated in advisory boards of MSD, BMS, Roche-Genentech, Janssen, Novartis, Bayer, Astellas. He has received research Funding form Roche-Genentech, Astellas, Pfizer. He has also received travel funding from Janssen, Merck, IPSEN, Pfizer. Clinical Trials: BMS, Roche-Genentech, Merck, EISAI, MSD, Gilead, Exelixis.