
Abstract
LRRK2 is a relatively common genetic risk factor for Parkinson’s disease (PD), with six coding variants known to cause familial PD. Non-coding variation at the same locus is also associated with sporadic PD. LRRK2 plays a role in many different intracellular signaling cascades including those involved in endolysosomal function, cytoskeletal dynamics, and Ca2+ homeostasis. PD-causing LRRK2 mutations cause hyperactive LRRK2 kinase activity, resulting in altered cellular signaling. Importantly, LRRK2 is lowly expressed in neurons and prominently expressed in non-neuronal cells in the brain. In this review, we will summarize recent and novel findings on the effects of PD-causing LRRK2 mutations in different nervous system cell types. This review will also provide novel insight into future areas of research at the intersection of LRRK2 cell biology, cell type specificity, and PD.
INTRODUCTION
Parkinson’s disease genetics
Parkinson’s disease (PD) is a progressive neurodegenerative disorder that affects approximately 1–2% of people over age 60 [1–5]. The number of affected individuals has more than doubled in the last 30 years, yet treatment options are currently focused on symptomatology rather than disease modification. The pathological hallmarks of PD are the progressive loss of neurons, prominently of dopamine (DA) neurons in the substantia nigra pars compacta (SNpc), and the presence of Lewy bodies and Lewy neurites composed of aggregated α-synuclein [3]. PD is clinically characterized by motor symptoms including bradykinesia, resting tremor, and muscular rigidity as well as non-motor symptoms that may predate the onset of motor symptoms and occur independently of dopaminergic neuron loss [3, 6].
The genetic underpinnings of PD are complex. PD was previously considered to be an entirely idiopathic disease because the origin and pathogenesis were unknown [7]. However, family studies have revealed monogenic forms of PD in which variation at a single gene locus can be attributed to disease causality [8]. For example, PD-causing autosomal dominant mutations have been identified in the LRRK2, SNCA, and VPS35 genes, and autosomal recessive mutations have been identified in the PARK7 (Dj-1), PRKN (Parkin), and PINK1 genes [7–11]. While the development of single gene disorders is primarily determined by genetics, known and unknown factors can modify disease penetrance and expressivity. For example, age is the greatest risk factor for both familial and sporadic PD [3, 12]. In the context of PD, highly penetrant causal variants, such as triplications of the SNCA gene, are rare, whereas variants with variable penetrance, such as those found at the LRRK2 or GBA loci, occur more frequently in the population [7, 10]. Adding to this complexity, PD-linked gene variants can also display variable expressivity, in which patients with the same pathogenic genotype may exhibit varying clinicopathological phenotypes [13–16]. Thus, age of onset, severity of illness, and pathology may be different across individuals with similar genetic background and family history.
Currently, monogenic PD accounts for approximately 5% of total cases and the other 95% fall into the category of sporadic PD [10]. However, even sporadic PD has polygenic influences on disease risk [7]. One method for narrowing down genetic risk for complex diseases are association studies such as genome-wide association studies (GWAS). GWAS compare the frequency of allelic variants between individuals with or without a given phenotype by analyzing large-scale population-level genotyping or sequencing data, thus identifying diseases-associated chromosomal regions or loci [17]. There are many auxiliary approaches to interpret GWAS signals, including expression quantitative trait loci (eQTL) analysis, in which gene expression data is paired with a reference GWAS to pinpoint genomic locations where gene expression segregates with allelic variation [18]. Follow up analyses can identify whether these eQTLs are context specific, i.e., linked to differences in gene expression in a specific tissue or cell type. Extensive functional experiments and very clear GWAS signal paired with supporting eQTL analyses are helpful to make the assertion that noncoding variation in a specific gene drives a disease phenotype. One very strong candidate gene that meets the above criteria for sporadic PD is LRRK2.
LRRK2 STRUCTURE AND FUNCTION
Beginning with the identification of the PARK8 locus on human chromosome 12 in a family from Sagamihara Japan, more than 100 coding variants have reported in LRRK2 [19, 20]. However, of these, only six coding variants have been reliably shown to be pathogenic by virtue of segregation in families, G2019S, I2020T, R1441C/G/H, and Y1699C [21–23]. There are also three other LRRK2 variants that do not have enough evidence to be disease-segregating, but are genetic risk factors for PD: I1372V, R1628P, and G2385R [24–27]. LRRK2 variants differ by population and ethnicity. For example, G2019S contributes to genetic risk in most countries, with the greatest prevalence in Ashkenazi Jewish and Berber populations, whereas R1628P and G2385R have only been found in Asian populations [25, 28]. Overall, Coding variants in LRRK2 are estimated to be found in ∼5% of familial and ∼1% of apparently sporadic PD cases [19]. Interestingly, age of onset and presentation of clinical symptoms for patients with LRRK2-linked monogenic PD are remarkably similar to those with sporadic PD, whereas other forms of monogenic PD tend to have clinically distinct phenotypes [21, 29–32].
Additionally, the region containing LRRK2 is an example of a pleomorphic risk locus, a location in the genome where both rare coding PD and common noncoding risk alleles associated with a similar disease coexist [33]. The chromosomal region including LRRK2 qualifies as this type of locus because the variants identified by GWAS are the LRRK2 p.G2019S coding mutation and a common noncoding variant (captured by the single nucleotide polymorphism rs76904798) that mediate disease risk independently of one another [34].
The LRRK2 gene encodes the protein leucine-rich repeat kinase 2 (LRRK2), a large (288 kDa) multimeric protein in the Roco family [35, 36]. LRRK2 has three protein-interactive domains towards its N-terminus: armadillo, ankyrin, and leucine-rich repeat motifs. Towards the C-terminus, LRRK2 has a WD40 domain preceded by a catalytic core composed of the kinase domain and a Ras-of-complex (ROC) GTPase domain that works in tandem with the C-terminal of Roc (COR) domain [35, 36]. The pathogenic LRRK2 coding variants are all found in the catalytic core: G2019S and I2020T are located in the kinase domain, R1441C/G/H are in the ROC domain and Y1699C is in the COR domain [37]. The ROC/COR domains of LRRK2 catalyze the hydrolysis of GTP to GDP [37]. LRRK2 also functions as a serine-threonine kinase capable of both autophosphorylation at its serine-1292 residue and phosphorylation of target substrates including Rab GTPases [37]. LRRK2 can also be phosphorylated by upstream or downstream proteins at constitutive phosphorylation sites (serines 860, 910, 935, 955 and 973/976) localized and clustered in the leucine-rich repeat domain which can provide insight into LRRK2 signaling pathways [38].
LRRK2 is expressed in peripheral organs including liver, kidney, heart, and lung, as well as in the brain with expression patterns varying by cell type [39–48]. In the mammalian brain, LRRK2 mRNA and protein are highly expressed in brain areas innervated by dopamine neurons; however, cell type specific expression varies by species [46–48]. In the human cortex and SNpc, LRRK2 mRNA expression is greater in microglia, excitatory neurons, and oligodendrocyte precursor cells than other cell types [45]. In contrast, in mice, LRRK2 expression is greater in astrocytes compared to microglia [44]. Compared to other PD-associated genes, there is very low LRRK2 mRNA and protein in dopamine neurons themselves [46].
The physiological role of LRRK2 has yet to be fully determined but growing evidence supports a role for LRRK2 in the endolysosomal system [49]. Under baseline conditions, LRRK2 is present in the cytosol of transfected cells [50]. However, we have shown that treating cells with lysosomal damaging agent, L-leucyl-L-leucine methyl ester (LLOMe), recruits LRRK2 to lysosomal membranes and activates LRRK2 kinase activity [51]. Similar effects are seen with a variety of lysosomal damaging agents [52–54]. Furthermore, LRRK2 can be recruited to different membranes in the endolysosomal system in which it’s then associated with phosphorylation of target proteins such as Rab10 and Rab12, and subsequent recruitment of adaptor proteins including c-Jun NH2-terminal kinase-interacting protein 4 (JIP4) to the same membranes [51, 56]. LRRK2 kinase activity mediates lysosome size, number, and degradative capacity and pathogenic PD-linked LRRK2 mutations have been shown to disrupt autophagic processes, which require lysosomal activity for completion [57]. For example, one study showed reduced lysosomal degradation, slower clearance of α-synuclein, and abnormal lysosomal clustering in the perinuclear region of mouse embryonic fibroblasts (MEFs) from LRRK2 p.R1441G knock-in mice [58]. The clustering phenotype was also observed in the aged striatum from mouse carrying LRRK2 mutations [58]. Additionally, many studies have documented autophagic, vesicle trafficking, and lysosomal defects in LRRK2 p.G2019S mutant astrocytes, neurons, and microglia [59]. These results imply that LRRK2 is an important regulator of endolysosomal function in the brain.
LRRK2 may also operate as a scaffold in different signal transduction pathways, utilizing its protein-interactive domains to tether proteins into configurations that facilitate pathway-relevant interactions and functions [60]. Screening of the LRRK2 interactome suggests that LRRK2 may be involved in pathways regulating cytoskeletal dynamics as LRRK2 interacts with actin as well as proteins involved in actin filament remodeling and regulation [61]. Knockdown of LRRK2 leads to significant morphological changes in murine fibroblasts and dopaminergic neurons, providing additional evidence that LRRK2 is important for healthy cytoskeletal function and maintaining typical cell structure [61]. Additionally, LRRK2 may act as a scaffold in the canonical Wingless-related Integration Site (Wnt) signaling pathway, connecting proteins in the b-catenin destruction complex (BDC) in the absence of Wnt stimulation and tethering the BDC to the membrane-bound Wnt receptor complex in the presence of Wnt [62]. This set of interactions has been supported by co-immunoprecipitation experiments showing interactions between LRRK2, lipoprotein receptor-related protein 6 (LRP6; a co-receptor for Wnt ligands), and multiple proteins in the BDC [62]. Furthermore, functional experiments showed weakened Wnt signaling in the context of PD-associated LRRK2 mutations or kinase inhibition [63].
LRRK2 has also been shown to regulate mitochondrial dynamics [64]. Mitochondria undergo fission and fusion events to adjust to changes in energy demand and to restore or remove damaged mitochondria as a form of quality control in response to cellular stress [65]. Overexpression of wildtype LRRK2 leads to kinase-dependent fission, upregulated recruitment of fission proteins, slowed fusion, increased reactive oxygen species (ROS) production, and decreased energy production in primary neurons [64]. Pathogenic LRRK2 variants, specifically p.G2019S and p.R1441C, have an even stronger effect of mitochondrial phenotypes [64].
The evidence supporting LRRK2 involvement in a variety of intracellular signaling pathways combined with its differential expression patterns across cell type and species suggest that LRRK2 function may have cell type specific effects. Here, we will summarize current research on the impact of pathogenic LRRK2 mutations on three major central nervous system (CNS) cell types: neurons, astrocytes, and microglia.
NEURONS
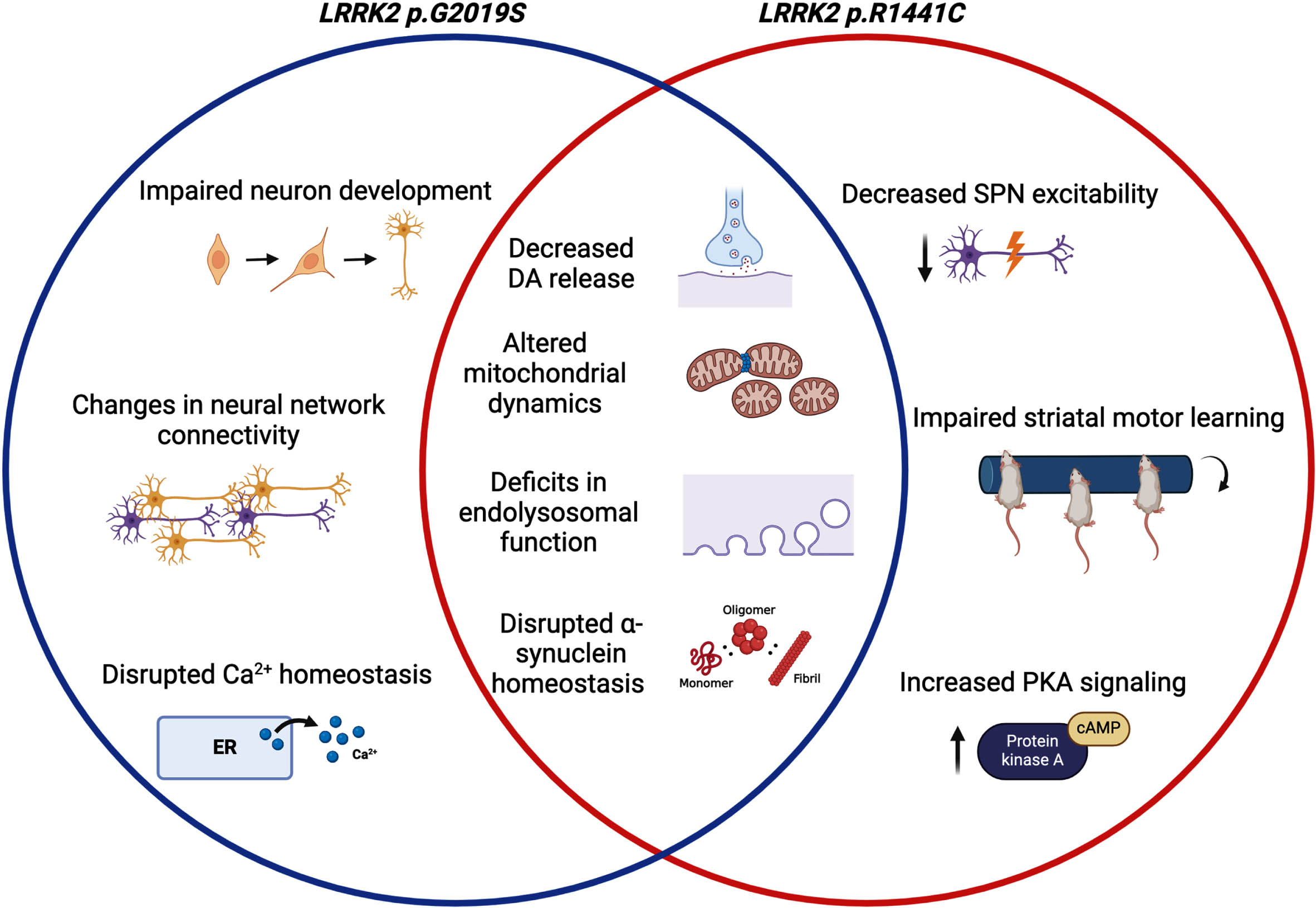
Neurons are perhaps the most obvious cell type to interrogate for mechanisms related to neurodegenerative diseases. However, unlike other PD genes of interest, endogenous LRRK2 expression is low in midbrain dopaminergic neurons, which are strongly affected in PD [45, 67]. In contrast, expression of LRRK2 is high within spiny projection neurons (SPNs) in the striatum that are the synaptic targets of SNpc dopamine neurons [67]. LRRK2 is also expressed in subsets of cortical neurons and in other brain regions [45, 67]. Here, we will discuss the various ways in which LRRK2 has been proposed to impact neuronal function, noting that it is likely that effects will vary across neuronal types and that non-cell autonomous effects of LRRK2 may represent important pathogenic mechanisms (as discussed below).
LRRK2 MUTATIONS CAN ALTER NEURONAL CONNECTIVITY AND EXCITABILITY
Given the high expression of LRRK2 in SPNs, several studies have examined how LRRK2 affects electrophysiology in these cells. Using voltammetry to measure current in striatal tissue of LRRK2 p.G2019S and p.R1441C knock-in mice, Xenias and colleagues found a decrease in evoked dopamine release in LRRK2 mutant mice compared to WT [68]. To further explore the impact of decreased dopamine release, Xenias et al. measured and compared the excitability of indirect and direct pathway SPNs across LRRK2 mutant and WT mice [68]. Interestingly, only indirect pathway SPNs in LRRK2 p.R1441C knockin mice had significantly decreased excitability [68]. In addition, disruption of dopamine signaling via dopamine D1 and D2 receptor antagonism during the rotarod training task significantly impaired striatal motor learning in LRRK2 p.R1441C mice but not p.G2019S or WT mice [68]. Increased synaptic protein kinase A (PKA) signaling observed in the LRRK2 p.R1441C mice but not in LRRK2 p.G2019S mice was shown to underlie the LRRK2 p.R1441C-specific motor deficits [68]. Thus, this study highlights a difference in neuronal circuitry and signaling between pathogenic LRRK2 mutations but did not resolve why there are such differences. It would be interesting to examine SPN excitability, dopamine signaling, and striatal motor learning in the other PD-causing LRRK2 mutations (p.I2020T, p. R1441G, p.R1441H, and p.Y1699C). Furthermore, it would be valuable to determine if changes in basal ganglia circuitry and motor learning are dependent on established biochemical correlates of mutations such as accumulation of phosphorylated RAB proteins.
Monogenic LRRK2-linked PD may have changes in neural network connectivity prior to onset of motor symptoms. Using high spatial and temporal resolution calcium imaging, Carola et al. tracked the activity and interactions of induced pluripotent stem cell (iPSC)-derived dopamine neurons at a single-cell level [69]. The DA neurons were differentiated from PD patient-derived iPSCs expressing the LRRK2 G2019S mutation. Statistical analysis of transfer entropy was used to compute functional connectivity between pairs of active neurons in the network of interest [69]. DA neurons derived from LRRK2 p.G2019S patients had a lower density of connections and larger functional communities (groups of strongly connected neurons) with an elevated propensity for excessive network synchrony events, whereas neurons from control lines had greater connection density and smaller functional communities that had weaker synchrony events [69]. Thus, the network electrophysiology was shown to be altered in the context of LRRK2 p.G2019S [69]. Functional connectivity also differed on a subpopulation level [69]. Networks of TH+ dopaminergic neurons generated significantly more extreme bursting events in the presence of LRRK2 p.G2019S compared to control [69]. In addition, Carola et al. showed that LRRK2 p.G2019S DA neurons released less DA than control neurons, and in an aged culture, had increased morphological changes indicative of neurodegeneration and lowered cell viability compared to controls [69]. Future studies could expand on this set of observations by investigating DA neuron electrophysiology and DA production in mixed cultures to determine if glial cells modify neuronal output. It would also be important to determine if gene correction of p.G2019S can reverse any observed DA neuron phenotypes.
LRRK2 MUTATIONS CAN INFLUENCE NEURONAL α-SYNUCLEIN BEHAVIOR
On a cellular level, synaptic transmission has been shown to mediate α-synuclein localization, release, and aggregation. Because LRRK2 mutations are associated with alterations in synaptic activity, multiple studies have explored the dynamics of neural transmission and α-synuclein aggregation in the context of PD-causing LRRK2 mutations [70, 71]. In addition, multiple studies have shown that neurons expressing the LRRK2 p.G2019S mutation have a tendency to accumulate α-synuclein puncta in the perinuclear region of cells [72–75]. For example, Wu et al. demonstrated that enhancing neuronal activity in WT murine brain slice cultures pretreated with α-synuclein preformed fibrils (PFFs) significantly increased α-synuclein aggregation and accelerated dopaminergic neurodegeneration compared to untreated slice cultures [71]. The injection of sonicated PFFs into cells is commonly used to model α-synuclein pathology observed in PD because it is thought to corrupt endogenous α-synuclein and make it more prone to forming pathological aggregations [71]. Interestingly, stimulating neural activity in PFF-treated LRRK2 p.G2019S slice cultures greatly exacerbated α-synuclein pathology and increased Lewy body formation when compared to WT PFF-treated cultures [71].
In a separate study, Volpicelli-Daley et al. found that primary mouse neurons from LRRK2- LRRK2 p.G2019S transgenic mice as well as dopaminergic neurons in the SNpc of LRRK2 p.G2019S transgenic rats sequester endogenous α-synuclein into inclusions in response to PFFs in a kinase-dependent manner [72]. Imaging and immunoblotting experiments showed that LRRK2 p.G2019S neurons had more endogenous α-synuclein in basal conditions and produced more inclusions upon PFF exposure compared to WT-LRRK2-overexpressing and non-transgenic neurons [72]. Furthermore, the proportion of membrane-associated α-synuclein to cytosol-associated α-synuclein was different in LRRK2 p.G2019S neurons in that more α-synuclein was in the cytosolic fraction than in non-transgenic neurons [72]. Monomeric soluble α-synuclein found in the cytoplasm is thought to be more prone to aggregation compared to membrane-bound oligomeric α-synuclein [76]. Because PD-linked SNCA mutations decrease the α-synuclein tetramer:monomer (T:M) ratio, a 2022 study looked into the effect of pathogenic LRRK2 mutations on α-synuclein multimerization [77]. A decreased T:M ratio has been shown to increase pSer129 α-synuclein hyperphosphorylation, have cytotoxic effects on neurons, and cause tremor and gait deficits in mice [77]. Using iPSC-derived excitatory cortical-like neurons, Fonseca-Ornelas et al. found that either LRRK2 p.G2019S or LRRK2 p.R1441C decreases the cytoplasmic α-synuclein T:M ratio and increases the phosphorylation of pSer129 α-synuclein monomers [77]. Conversely, LRRK2 kinase inhibition restored the α-synuclein T:M ratio and phosphorylation of endogenous α-synuclein to WT levels in LRRK2 mutant neurons [77]. The same study then showed a kinase-dependent decrease in the α-synuclein T:M ratio in iPSC-derived midbrain dopaminergic neurons expressing LRRK2 p.G2019S [77]. Thus, these results suggest that mutant LRRK2 hyperactive kinase activity disrupts α-synuclein homeostasis in PD-relevant cell subtypes. Importantly, these studies provide a link between LRRK2 and α-synuclein multimerization and suggest a possible common avenue of pathogenesis linking these two important genes for PD.
LRRK2 MUTATIONS DISRUPT ENDOLYSOSOMAL FUNCTION IN NEURONS
Pathogenic LRRK2 mutations have been shown to disrupt endolysosomal membrane trafficking of proteins, including α-synuclein, in neurons [78]. For example, Brzozowski and colleagues found that LRRK2 influences anterograde axonal transport of α-synuclein to the presynaptic terminal [70]. In vitro and in vivo live imaging and biochemical experiments revealed that LRRK2 knockout or kinase inhibition increases anterograde axonal transport of α-synuclein to the presynaptic terminal of glutamatergic, nigrostriatal, and corticostriatal neurons [70].
In a separate study investigating LRRK2-mediated axonal transport, Boecker and Holzbaur showed that the presence of the LRRK2 p.G2019S mutation significantly disrupted retrograde axonal autophagosome transport in a kinase-dependent manner due to the increased frequency of autophagosome pausing and reversing direction [79]. In a healthy neuron, autophagosomes are formed in the axon and transported in a retrograde manner to the soma by motor protein, dynein [79]. In addition to dynein, the motor protein that moves transport in the opposing anterograde direction, kinesin, is also bound to autophagosomes but resides in an inhibited state [79]. During transport, autophagosomes fuse with lysosomes and become mature autolysosome organelles with degradative ability [79]. The dysregulation of autophagosome transport was observed in 3 separate models: rat hippocampal neurons overexpressing LRRK2 p.G2019S, cortical neurons cultured from LRRK2 p.G2019S knock-in mice, and excitatory cortical neurons differentiated from a LRRK2 p.G2019S hiPSC line [79]. Mechanistically, Boecher and Holzbaur showed that LRRK2 p.G2019S hyperactive kinase activity leads to the recruitment and phosphorylation of Rab proteins and JIP4 at the autophagosomal membrane [79]. Activation of JIP4 then causes abnormal activation of kinesin-1, which may lead to altered axonal transport due to a tug-of-war between competing motors acting on the autophagosome [79]. Overall, this suggests that LRRK2 p.G2019S hyperactive kinase activity ultimately causes autophagy impairment in neurons by disrupting autophagosome transport and preventing autophagosome maturation.
Clathrin-mediated endocytosis from the plasma membrane is also disrupted in the presence of pathogenic LRRK2 mutations. Dopaminergic neurons carrying the LRRK2 p.G2019S variant have a decrease in the number of synaptic vesicles paired with a heightened accumulation of clathrin-coated vesicles at the synapse [80]. Clathrin and adaptor protein complex 2 (AP2) coat membrane-bound cargo and begin the clathrin-mediated endocytic cycle [81]. Liu and colleagues demonstrated that LRRK2 binds and phosphorylates the M1 subunit of the AP2 complex (AP2M1) in vitro and in vivo [81]. Phosphorylation of AP2M1 is important for AP2M1 membrane association and initiating clathrin-coated vesicle formation [81]. Dephosphorylation of AP2M1 following vesicle scission is necessary to remove AP2M1 from the clathrin-coated vesicle, allowing for a new cycle of vesicle formation to begin [81]. LRRK2 knockout mouse brains were shown to have significantly lower levels of endogenous AP2M1 phosphorylation compared to WT, whereas LRRK2 p.G2019S knockin mouse brains had significantly higher AP2M1 phosphorylation compared to WT [81]. In the presence of LRRK2 p.G2019S, LRRK2-mediated AP2M1 phosphorylation exacerbates the association between AP2M1 and the plasma membrane and impairs endocytosis in neurons [81]. Using an in vitro uncoating assay, Liu et al. showed that hyperactive kinases activity exhibited by LRRK2 mutants inhibits the uncoating of AP2M1 from clathrin-coated vesicles [81]. It was also shown that overexpression of AP2M1 increases dopaminergic neuron loss in drosophila overexpressing WT or mutant LRRK2 [81]. Taken together, these findings indicate that hyperactive LRRK2 kinase activity impairs clathrin-mediated endocytic trafficking in dopaminergic neurons.
In a separate study, Bono et al. demonstrated that the presence of LRRK2 p.G2019S inhibits the trafficking of dopamine D3 receptors (D3Rs) and nicotinic acetylcholine receptors (nAChRs) to the membranes of hiPSC-derived dopaminergic neurons as well as the formation of the D3R-nAChR heteromer [82]. Activation of nAChRs or D3Rs leads to formation of the D3R-nAChR heteromer, which subsequently promotes the neurotrophic morphological remodeling of DA neurons [82]. Specifically, chronic stimulation of nAChRs or D3Rs by agonists such as nicotine or ropinirole, respectively, increases dendrite length, dendritic arborization, and soma area in iPSC-derived DA neurons [82]. In LRRK2 p.G2019S neurons, the neurotrophic effects of nicotine and ropinirole were nearly abolished and both D3Rs and nAChRs localized to the golgi apparatus instead of the membrane [82]. Treatment with LRRK2 kinase inhibitor, GSK2578215A, restored the neurotrophic function of nicotine and ropinirole and the heteromization of D3R and nAChR, implying that one or more LRRK2 substrates were responsible for these effects [82].
Neuronal autophagy and lysosomal function are two other endolysosomal processes impacted by pathogenic LRRK2 variants. Wallings et al. demonstrated that overexpression of WT or LRRK2 p.G2019S inhibited autophagosome production in primary rat cortical neurons, whereas overexpression of LRRK2 p.R1441C LRRK2 increased lysosomal pH, decreased autolysosome maturation, and diminished autophagosome-lysosome fusion [83]. Follow up in vivo experiments confirmed disrupted autophagic function in LRRK2 mutant neurons [83]. Aged (22 month) transgenic rats expressing LRRK2 p.R1441C had significantly greater LC3 puncta, an autophagosome and autophagy marker, in midbrain dopaminergic and cortical neurons than WT animals [83]. Interestingly, aged transgenic rats expressing LRRK2 p.G2019S had increased LC3 puncta only in midbrain dopaminergic neurons, suggesting that LRRK2 mutations may differentially affect neuronal subpopulations [83]. The same study then investigated the role of LRRK2 in lysosomal pH regulation in primary rat cortical neurons. The v-type H+ ATPase proton pump (vATPase) a1 subunit is enriched in cortical neurons and is trafficked back and forth between the Golgi to the lysosome where it forms a complex able to regulate lysosomal pH [83]. Interactions between vATPase a1 and its binding partners facilitates its localization and trafficking [83]. In neurons, vATPase a1 was significantly downregulated in LRRK2 p.R1441C neurons and upregulated in LRRK2 p.G2019S cortical neurons [83]. Wallings et al. went on to identify an interaction between LRRK2 and the a1 subunit of the in cortical neurons expressing WT or LRRK2 p.G2019S [83]. Expression of either LRRK2 mutations (LRRK2 p.G2019S or p.R1441C) led to dysregulated trafficking and localization of vATPase a1 [83]. In LRRK2 p.G2019S cortical neurons, vATPase a1 protein expression was greatly increased in the cellular lysosomal fraction and decreased in the Golgi fraction [83]. In contrast, the presence of the LRRK2 p.R1441C mutation abolished the interaction between LRRK2 and a1 leading to a significant decrease of vATPase a1 protein expression in both Golgi and lysosomal fractions [83]. Treatment with clioquinol, a zinc/copper ionophore, led to the zinc-dependent rescue of the observed LRRK2 p.R1441C-mediated phenotypes suggesting that zinc modulation may improve lysosomal health and function [83].
LRRK2 MUTATIONS CAN DISRUPT NEURONAL Ca2+ HOMEOSTASIS
Outside of the endolysosomal system, LRRK2 mutations have been shown to interfere with Ca2+ homeostasis and mitochondrial function. One study investigated the impact of LRRK2 p.G2019S on ER Ca2+ regulation using iPSC-derived neurons [84]. Korecka et al. found that pharmacological inhibition of the Sarcoendoplasmic Reticulum Calcium ATPase (SERCA) to inhibit ER Ca2+ influx resulted in significantly decreased neurite length in LRRK2 p.G2019S iPSC-derived neurons [84]. SERCA inhibition also increased depolarization-induced Ca2+ influx, suggesting that LRRK2 p.G2019S neurons have decreased Ca2+ buffering capacity [84]. LRRK2 p.G2019S iPSC-derived neurons showed delayed outgrowth of neurites compared to wild type cells [84]. This phenotype could be rescued by LRRK2 kinase inhibition when cells were treated with low doses (10 nM) of SERCA inhibitor, but it did not rescue more potent (100 nM) SERCA inhibition [84]. In addition, live calcium imaging demonstrated that LRRK2 p.G2019S neurons had lower levels of ER Ca2+ compared to WT iPSC-derived neurons [84]. RNA sequencing of LRRK2 p.G2019S iPSC-derived neurons and their isogenic mutation-corrected controls revealed significant downregulation of STIM1, ORAI1, and TRPC1, genes necessary for store-operated Ca2+ entry (SOCE) [84].
Furthermore, in a 2021 study, ribosome profiling revealed differential expression of genes involved in Ca2+ homeostasis when comparing iPSC-derived DA neurons from PD patients with LRRK2 p.G2019S relative to wild type cells [85]. Follow up experiments showed that LRRK2 p.G2019S DA neurons have increased intracellular Ca2+ concentration and amount of Ca2+ currents compared to control [85]. Wren Kim and colleagues found that adeno-associated virus (AAV)-mediated overexpression of a mutant non-phosphorylatable form of ribosomal protein, S15, restored intracellular Ca2+ concentration and altered Ca2+ currents in LRRK2 p.G2019S DA neurons to WT levels [85]. This is in line with previous research showing that phosphorylated S15 promotes hyperactive protein synthesis in G2019S LRRK2 neurons [85]. Furthermore, treating LRRK2 p.G2019S DA neurons with an L-type voltage gated Ca2+ channel (VGCC) antagonist also rescues Ca2+ homeostasis [85]. L-type VGCCs allow for the continuous Ca2+ influx during autonomous pacemaking in SNpc DA neurons and therefore, suppression decreases Ca2+ influx into the cell [85]. Similarly, translating ribosome affinity purification (TRAP) sequencing allowed Pallos et al. to identify the differential expression of genes involved in Ca2+ homeostasis between WT and transgenic dopaminergic neurons expressing LRRK2 p.G2019, as well as genes relevant to DNA repair, mRNA metabolism and mRNA translation [86].
Mutant LRRK2-mediated disruptions in Ca2+ signaling can also lead to altered immune signaling in neurons. Interferon-γ (IFN-γ) treatment has been shown to upregulate LRRK2 mRNA expression in iPSC-derived DA neurons [87]. A 2020 study demonstrated that LRRK2 operates downstream of IFN-γ in the IFN-γ signaling pathway and LRRK2 p.G2019S alters neuronal intracellular response to IFN-γ [88]. Specifically, the presence of LRRK2 p.G2019S and subsequent increase in LRRK2 protein levels inhibits the translocation of nuclear factor of activated T-cells (NFAT) to the nucleus via alterations in Ca2+ signaling and microtubule organization as well as significantly decreases AKT serine/threonine kinase 3 (AKT3) phosphorylation when compared to control iPSC-derived DA neurons [88]. NFAT nuclear translocation is crucial for inducing the transcription of cytokines and other immune response genes, thus the presence of LRRK2 p.G2019S likely alters neuronal immune response and could potentially contribute to disease pathology [88].
LRRK2 MUTATIONS CAN ALTER NEURONAL MITOCHONDRIA DYNAMICS
Wang et al. demonstrated that LRRK2 alters mitochondrial dynamics in SH-SY5Y neuroblastoma cells and neurons [64]. Electron and laser microscopy were used to show that mitochondria in WT SY5Y cells were tubular and filamentous whereas cells overexpressing WT LRRK2 had mitochondrial fragmentation and slower mitochondrial fusion events [64]. Fragmentation and slowed fusion were exacerbated in cells transfected with LRRK2 p.R1441C or p.G2019S, which had significantly more dynamin-like protein 1 (DLP1) recruitment to mitochondria [64]. Co-immunoprecipitation and fractionation experiments suggested that LRRK2 and DLP1 directly interact in the mitochondria, with stronger interaction between DLP1 and LRRK2 in LRRK2 p.G2019S and LRRK2 p.R1441C cells compared to WT and control [64]. In addition, overexpression of a non-functional DLP1 mutation reversed the effects of LRRK2 and LRRK2 variants on mitochondrial fragmentation, mitochondrial dysfunction, and cellular vulnerability to toxins and oxidative stress [64]. Validating the results seen in the SH-SY5Y cells, mitochondrial fractionation was increased in primary rat cortical neurons transfected with WT LRRK2 and further increased in neurons transfected with LRRK2 p.G2019S or LRRK2 p.R1441C [64]. Cell viability also drastically declined in WT-LRRK2 and mutant-LRRK2 transfected neurons, however co-transfection with the non-functional DLP1 mutant completely alleviated primary cortical neuron cell death [64]. These results suggest that LRRK2 and DLP1 both play crucial roles in regulation of mitochondrial fission and dysregulation of these proteins can exert mitochondria-mediated cytotoxic effects [64].
LRRK2 MUTATIONS AND NEURAL DEVELOPMENT
Despite the age-dependent penetrance of PD, some studies have investigated how LRRK2 mutations may alter neuronal development. Walter et al. measured changes in development over time by collecting cells at time points ranging from 4 to 42 days after inducing neural differentiation in human-derived neuroepithelial stem cells (NESCs) [89]. Prior to differentiation, RNAseq analysis revealed an upregulation of midbrain-dopaminergic-neuron-specific transcripts in LRRK2 G2019S stem cells compared to controls [89]. Imaging, flow cytometry, and RNAseq analysis showed that LRRK2 G2019S NESCs differentiated into neurons and gained midbrain dopaminergic specificity faster than cells from healthy or control lines [89]. However, this accelerated development was also accompanied by increased apoptosis [89]. These cells also lost proliferative ability and stemness faster than isogenic or healthy controls [89]. Interestingly, this same lab previously investigated the impact of the LRRK2 p.R1441G mutation on neural development in primary neural stem cells and observed impaired neuron differentiation and increased cell death [90].
A separate study generated iPSC lines from monogenic LRRK2 p.G2019S PD patients, sporadic PD patients, and healthy, age-matched (but not isogenic) controls, then differentiated them into ventral midbrain dopaminergic neurons [91]. In the short-term, dopamine neurons derived from all genotypes (PD and control) were phenotypically identical [91]. However, long-term culturing (>75 days) revealed the spontaneous development of distinct morphological alterations in DA neurons derived from both LRRK2-related PD and sporadic PD patients [91]. Control DA neurons showed mature morphology with long neurites and complex arborization, whereas DA neurons derived from Parkinson’s patients took on either a more immature phenotype with fewer neurites and less complex arborization or a neurodegenerative phenotype in which neurons had short or missing neurites, soma vacuolation, fragmented nuclei, and positive staining for cleaved caspase 3 (an apoptotic protein) [91]. An interesting expansion on this study would be to observe if the morphological changes that arise from long-term culturing of iPSC-derived neurons reflect DA neuronal morphology in brain tissue from individuals carrying the LRRK2 p.G2019S mutation, and therefore accurately recapitulate adult neuron morphology.
Collectively, these various reports of neuronal phenotypes in cells and tissues with LRRK2 mutations, suggest that neurons may be a critical cell type for the expression of LRRK2-associated disease. However, several important questions remain to be answered, particularly in the relationship of neuronal dysfunction with the known signaling pathways related to LRRK2 substrates. Additionally, most studies have focused on one or two mutations and so it remains to be seen whether all of the above phenotypes are congruent across all pathogenic variants, which would support that neuronal dysfunction contributes to disease. Irrespective of such considerations, it should be noted that LRRK2 is also expressed in non-neuronal cells, and thus non-cell autonomous mechanisms may also contribute to disease pathophysiology. The low expression of LRRK2 in SNpc dopaminergic neurons might also support that other cell types could contribute to neuronal cell death in LRRK2-associated PD, although a reasonable counter-argument is that low expression might reflect a high sensitivity of dopamine neurons to excess LRRK2 activity. Thus, it will be crucial for future studies into LRRK2-mediated DAergic neurodegeneration to expand from a single cell lens to a multicellular view that takes changes in intercellular communication into account.
ASTROCYTES
Astrocytes are a subset of glial cells that tile all brain regions in a highly organized manner [92, 93]. Separated into two distinct biochemical and developmental categories, protoplasmic astrocytes are found in gray matter and fibrous astrocytes line white matter tracts in the brain [94]. Astrocytes are crucial to the cellular ecosystem of the CNS [95]. Astrocytes are involved in the formation, maintenance, and pruning of neuronal synapses, the regulation of neural blood flow, and the maintenance of the blood -brain barrier (BBB) [93]. They can also release gliotransmitters and growth factors to stimulate short-term neural activity or facilitate long-term synapse changes [93]. Considering the wide-ranging functions that these cells mediate, astrocytic dysfunction could potentially wreak on CNS homeostasis and communication, and astrocytes have become a key cell type to study in neurological disease pathogenesis.
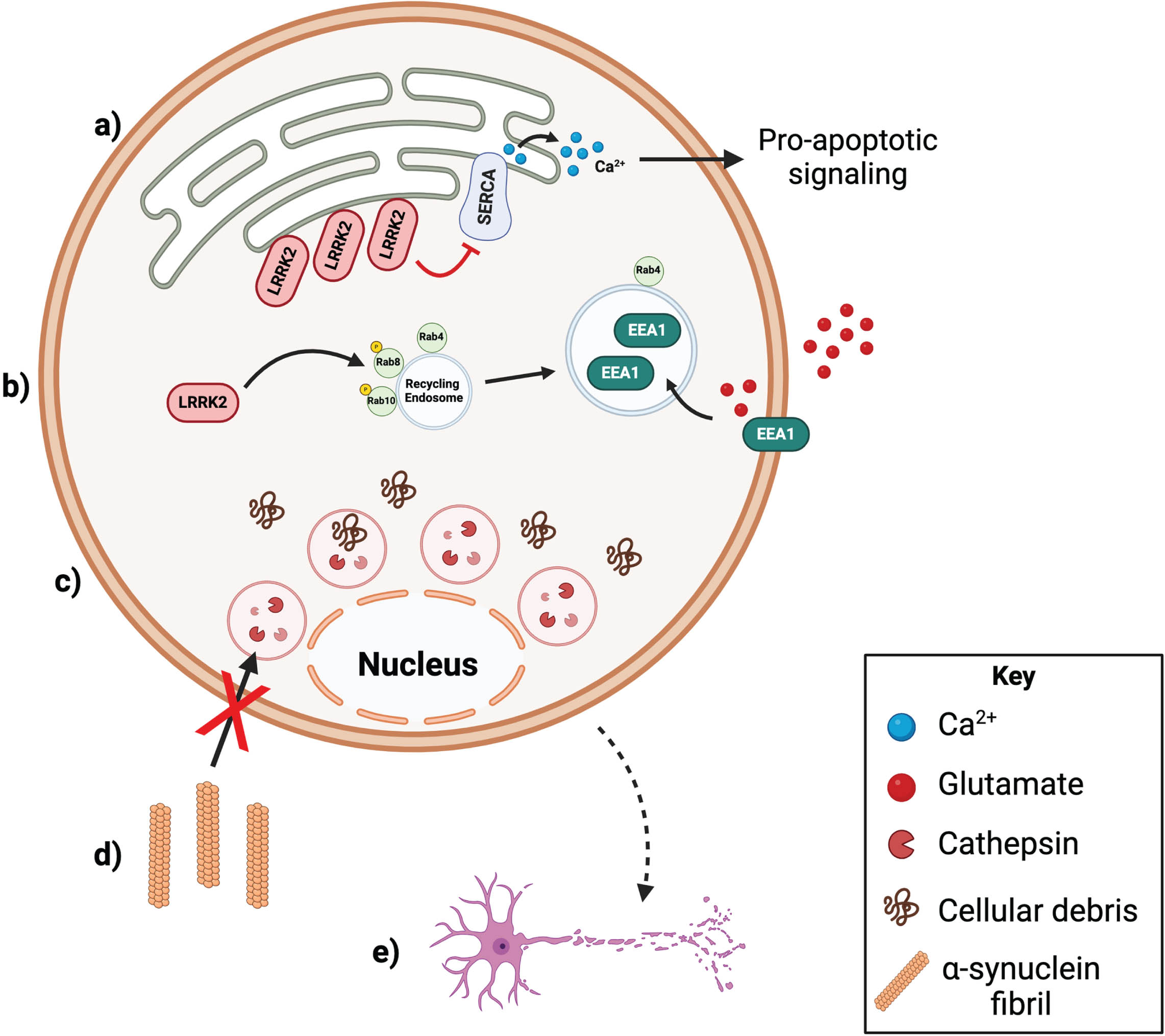
LRRK2 MUTATIONS CAN ALTER Ca2+ HOMEOSTASIS IN ASTROCYTES
LRRK2 is expressed in both murine and human astrocytes [44, 45]. It is therefore important to explore how LRRK2 variants may affect astrocytic function as a possible way to influence risk and/or pathogenesis of PD. In one study, Lee and colleagues assessed endoplasmic reticulum (ER) stress response in primary cortical astrocytes from LRRK2 p.G2019S knockin mice [96]. First, they found that LRRK2 p.G2019S astrocytes had significantly greater apoptotic cell death following treatment with ER-stress-response-inducing agents including oligomeric α-synuclein or tunicamycin [96]. Then, through a combination of techniques, the authors demonstrated that the LRRK2 p.G2019S mutation disrupts LRRK2 binding with a specific subset of 14-3-3 proteins, increasing LRRK2 recruitment to the ER membrane [96]. Once at the ER, LRRK2 p.G2019S directly interacts with and inactivates the sarcoendoplasmic reticulum Ca2+ ATPase (SERCA). LRRK2 tethers SERCA to phospholamban, inactivating SERCA and preventing Ca2+ reuptake [96]. Prolonged ER Ca2+ depletion and ER stress response results in a shift to proapoptotic signaling ultimately leading to astrocytic and eventually neuronal cell death [96]. Previous studies have also demonstrated a direct interaction between LRRK2 and 14-3-3 proteins and reported that some PD-causing mutations interfere with LRRK2 and 14-3-3 binding [97–100]. However, there are inconsistent and conflicting results regarding the effects of LRRK2 p.G2019S on 14-3-3 binding across cell types and assays [99]. Future research is necessary to develop stronger conclusions regarding the effects of PD-linked LRRK2 mutants on LRRK2-14-3-3 binding across different cell types.
LRRK2 MUTATIONS CAN DISRUPT ASTROCYTIC PROTEIN CLEARANCE AND DEGRADATION
Astrocytes play a critical role in regulating the concentration of proteins released into the synaptic space [93]. Astrocytic protein clearance relies on a functional endolysosomal system for efficient degradation of unwanted proteins and prevention of toxic protein aggregation. One study demonstrated that pathogenic LRRK2 mutations (LRRK2 p.G2019S, p.Y1699C, p.R1441C) have kinase-dependent effects on lysosomal morphology and degradative capacity in primary mouse astrocytes [57]. Henry et al. observed that astrocytes expressing mutant LRRK2 had significantly enlarged lysosomes arranged in a tight, perinuclear distribution [57]. Further, the number of lysosomes was significantly decreased in LRRK2 p.G2019S astrocytes, whereas the quantity of lysosomes nearly doubled in LRRK2 KO astrocytes compared to WT [57]. Additionally, lysosomal degradative function, Cathepsin B activity, and lysosomal pH were all significantly decreased in LRRK2 p.G2019S astrocytes [57]. LRRK2 kinase inhibition rescued the lysosomal defects and phenotypic changes observed in astrocytes expressing pathogenic LRRK2 mutations, demonstrating that LRRK2 affects lysosomes in astrocytes in a kinase-dependent manner [57].
To give a further example of how astrocytes control protein turnover, Streubel-Gallasch et al. found that primary mouse astrocytes carrying the LRRK2 p.G2019S mutation had a reduced ability to clear α-synuclein from the extracellular space [43]. Cortical and striatal astrocytes can internalize sonicated α-synuclein preformed fibrils and direct them to lysosomal-associated membrane protein 2A (Lamp2a)-positive endolysosomal organelles [43]. However, G2019S astrocytes internalized fewer α-synuclein particles than WT or LRRK2 KO astrocytes [43]. In addition, there were fewer lysosome-like structures, as well as altered endolysosomal vesicle morphology, in striatal astrocytes isolated from G2019S transgenic mice [43]. This group also found that LRRK2-mediated astrocytic α-synuclein clearance is LRRK2 kinase- and annexin A2 (AnxA2)-dependent [43]. AnxA2 is a phospholipid-binding protein almost exclusively expressed in glial cells and it’s known that AnxA2 deficits lead to impaired endocytic capacity [43]. AnxA2 levels are significantly decreased in both untreated and α-synuclein PFF-treated LRRK2 p.G2019S astrocytes [43]. Furthermore, siRNA knockdown of AnxA2 in WT astrocytes led to significantly decreased internalization of α-synuclein, confirming that AnxA2 loss of function impairs endocytosis [43]. LRRK2 kinase inhibition also restores AnxA2 levels and function [43]. Thus, the activity induced by pathogenic LRRK2 mutations decreases AnxA2 levels and ultimately leads to diminished α-synuclein clearance [43]. Based on these observations, it would be enlightening for future research to explore how overexpression or siRNA knockdown of AnxA2 alters α-synuclein behavior in an astrocyte-neuron co-culture model and across different LRRK2 genotypes.
Maintaining glutamate homeostasis is another mechanism by which astrocytes utilize the endolysosomal system to maintain the synapse and exert critical neuroprotective effects [101]. Glutamate is an abundant excitatory neurotransmitter essential for communication between cells in many neural circuits including the basal ganglia circuitry that dictates voluntary movement [102]. Astrocytes express the majority of the highly active glutamate transporter in the brain, excitatory amino acid transporter 2 (EAAT2) in humans and glutamate transporter 1 (Glt-1) in mice [103]. Dynamic and constitutive endocytic recycling of EAAT2 allows for the rapid, activity-dependent turnover of EAAT2 at the plasma membrane [104]. Specifically, one study showed significantly decreased EAAT2 protein (but not mRNA) expression and increased levels of glial fibrillary acidic protein (GFAP), a marker of reactive astrogliosis, in human postmortem midbrain tissue samples from LRRK2 p.G2019S familial PD patients, but not from sporadic PD patients and healthy controls [105]. Similarly, LRRK2 p.G2019S knock-in mice also had downregulated Glt-1 glutamate transporter signal and increased GFAP compared to WT animals [105]. The same study characterized the mechanism by which LRRK2 p.G2019S disrupts EAAT2 trafficking to the plasma membrane using primary striatal astrocytes and organotypic slices [105]. Iovino and colleagues also showed that LRRK2 p.G2019S causes enlargement of Rab4-positive fast recycling endosome by phosphorylating and inactivating both Rab8A and Rab10 [105]. In addition, the presence of LRRK2 p.G2019S leads to the preferential uptake of Glt-1 into Rab4-positive fast-recycling endosomes. Glt-1 also accumulates in Rab4-positive vesicles in WT astrocytes when treated with Monensin, a pharmacological inhibitor of endocytic recycling and activator of LRRK2 kinase activity [52, 105]. Furthermore, pharmacological inhibition of LRRK2 p.G2019S kinase activity with MLi-2 treatment led to reduced colocalization of Glt-1 with Rab4, corrected redistribution of Glt-1 at the plasma membrane, and restored Glt-1 glutamate uptake activity [105]. Collectively, these data indicate that LRRK2 p.G2019S may contribute to impaired glutamate homeostasis by disrupting Rab-mediated recycling of EEAT2 glutamate transporters to the membrane and preventing proper glutamate clearance. It would be interesting to learn if PD-linked LRRK2 mutations impair astrocytic uptake and/or recycling of neurotransmitters other than glutamate.
LRRK2 MUTATIONS CAN DISRUPT ASTROCYTE-MEDIATED INTERCELLULAR SIGNALING
Outside of lysosomal degradation and endocytic recycling, astrocytes also require a functional endolysosomal system to maintain critical communication with neurons. Co-culture studies using neurons and astrocytes generated from patient-derived iPSCs studies have shown that presence of the LRRK2 p.G2019S variant disrupts astrocyte-neuron interactions and contributes to neurodegenerative phenotypes such as less complex and shortened neurite morphology, abnormal α-synuclein accumulation, and decreased cell survival rates [106, 107]. Specifically, these studies have shown that co-culturing astrocytes carrying the LRRK2 p.G2019S mutation with WT neurons leads to the selective degeneration and loss of dopaminergic neurons, suggesting a non-cell autonomous effect of the mutation on neuronal survival [106, 107]. The selective loss of dopaminergic neurons has also been observed when WT neurons were cultured in LRRK2 p.G2019S astrocyte-conditioned media [106, 107]. Furthermore, co-culturing DA neurons expressing LRRK2 p.G2019S with WT astrocytes or WT astrocyte-conditioned media, partially restored neurodegenerative phenotypes [106, 107]. To better understand the mechanism underlying the astrocyte-mediated neurotoxicity observed in co-culture, de Rus Jacquet et al. characterized vesicular changes in LRRK2 p.G2019S and WT hiPSC-derived astrocytes [106]. Compared to isogenic and non-isogenic control iPSC-derived astrocytes, multivesicular bodies in the mutant LRRK2 lines had altered morphology and spatial distribution, being significantly smaller and further from the nucleus [106]. Furthermore, there was an increased accumulation of LRRK2 and ratio of phosphorylated α-synuclein to total α-synuclein in the multivesicular bodies of the LRRK2 p.G2019S astrocytes [106]. Enzyme-linked immunosorbent assay (ELISA) and co-culture experiments confirmed the presence of LRRK2 and α-synuclein in secreted extracellular vesicles and that dopaminergic neurons uptake extracellular vesicles secreted by the iPSC-derived-astrocytes [106]. In a separate study, di Domenico et al. focused on the autophagic mechanisms underlying the astrocyte-mediated α-synuclein aggregation in neurons [107]. Di Domenico and colleagues observed decreased chaperone mediated autophagy (CMA), defects in protease-mediated α-synuclein degradation, and impaired macroautophagy [107]. In contrast, treating a coculture of WT DA neurons and LRRK2 p.G2019S astrocytes with a CMA activator greatly decreased α-synuclein accumulation and partially rescued neuron survival and morphology [107].
MUTANT LRRK2 CAN INDIRECTLY ALTER BBB INTEGRITY
Astrocytes regulate blood vessel formation, blood flow, and overall BBB health via secreted factors and direct interactions with other cell types in the neurovascular unit including endothelial cells and pericytes. Because astrocytic dysfunction and increased neuroinflammation are commonly observed in PD patients, a 2023 study investigated potential links between astrocyte-mediated immune response, LRRK2, and BBB integrity. de Rus Jacquet et al. generated a 3D co-culture model of the BBB composed of a microfluidic chip containing a top lane with iPSC-derived endothelial cells to represent the vascular compartment, a middle scaffold lane composed of extracellular matrix gel, and a bottom lane with iPSC-derived astrocytes and primary human pericytes representing the brain compartment. When the 3D BBB chip contained iPSC-derived astrocytes with the LRRK2 p.G2019S mutation, endothelial cell morphology was altered and there was significantly more leakage of fluorescent dyes into the brain compartment than in 3D BBB chips with control iPSC-derived astrocytes. Furthermore, gene expression of proinflammatory factors secreted by astrocytes, including interleukin-6 and interleukin-8 that are known to mediate vascular function and BBB permeability, were shown to be upregulated in LRRK2 p.G2019S astrocytes. Computational analysis identified the mitogen-activated protein kinase (MEK)/extracellular signal-regulated kinase (ERK) signaling cascade as a LRRK2 target and a modulator of angiogenesis and inflammatory response. The same study went on to show that MEK1/2 inhibition rescued the pro-inflammatory astrocytic phenotype and restored endothelial barrier integrity in the 3D BBB chips containing LRRK2 p.G2019S astrocytes. The results from the 3D BBB chip model recapitulates MRI and PET data from other studies showing increased capillary leakage into the midbrain of PD patients as well as replicates the morphological changes in endothelial vasculature observed in postmortem SNpc tissue from PD patients.
Collectively, these observations demonstrate that LRRK2 mutations affect astrocyte function, particularly for mouse astrocytes in vitro and in vivo. It is very likely that the underlying mechanism relates to altered endolysosomal function, and the current literature supports the idea that many of the effects of LRRK2 mutations in astrocytes drive non-cell autonomous effects on neurons.
MICROGLIA
Microglia are the resident macrophages of the CNS whose primary functions include phagocytosis, surveillance of the cellular environment, and initiation of signaling cascades [108]. Through these functions, microglia contribute to many larger cellular processes such as inflammatory response, synaptic remodeling, and maintenance of neuronal health [108]. While previous literature has often categorized microglia into resting vs. activated cells, it is currently agreed that microglia are highly dynamic cells that can attain multiple biochemical and morphological states in response to different contexts including neuroanatomical location, age, species, and disease [108]. With age, microglia have a heightened response to stimulation and a reduced sensitivity to regulating molecules [109]. Thus, microglia are an important cell type to study in the context of PD because overactive microglial response, and upregulated proinflammatory signaling are common pathological features of PD [110].
GWAS analysis and RNA sequencing studies have been key tools for understanding how microglial behavior and immune response is altered across different PD-relevant contexts. Access to human data is essential for addressing any potential species differences between rodent and human cells. For the current discussion, it is important to note that human microglia have greater spatial and temporal diversity than rodent microglia, as well as having differences in protein expression [111]. For example, LRRK2 expression is minimal in rodent microglia, but highly expressed in the human brain [44, 45]. Conversely, toll-like receptor 4 (TLR4), a microglial receptor that initiates pro-inflammatory signaling, is highly expressed in the rodent brain, but has relatively low expression in the human brain [111].
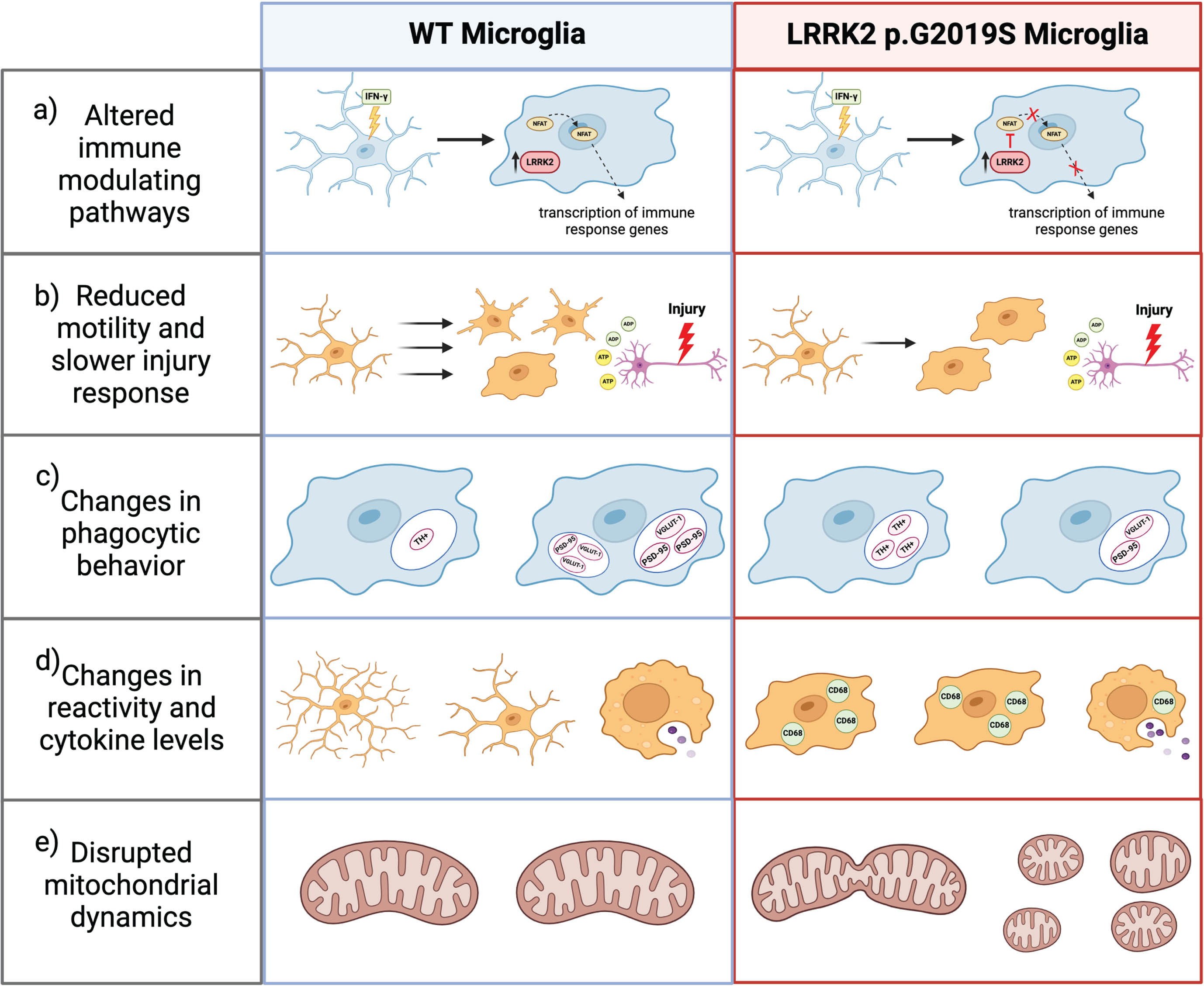
LRRK2 MUTATIONS CAN MODULATE IMMUNE RESPONSE AND UPREGULATE PROINFLAMMATORY SIGNALING
PD-linked LRRK2 mutations have been shown to modulate the gene expression profile of microglia and the immune response in the central nervous system and periphery [112, 113]. Expression studies have shown that immune cells in the periphery release greater concentrations of inflammatory cytokines and have higher levels of LRRK2 in PD patients compared to age-matched controls [112]. Within the CNS, LRRK2 p.G2019S carriers have greater microglial activation in the SNpc regardless of whether they presented PD symptoms compared to controls [114]. A separate study showed that young and aged LRRK2 p.G2019S transgenic mice have markedly higher protein levels of CD68 (cluster of differentiation 68), a marker of microglial activation, replicating the results seen in human G2019S carriers [115]. Furthermore, mice carrying the LRRK2 p.G2019S mutation show an age-related increase in mRNA of pro-inflammatory molecules, tumor necrosis factor α (TNF-α), inducible nitric oxide synthase (iNOS), interleukin 6 (IL-6), and IFN-γ, in the prefrontal cortex (PFC) and IFN-γ in the striatum [115]. Other studies have shown that LRRK2 p.G2019S also promotes greater secretion of proinflammatory cytokine, TNF-α, by primary rodent microglia and in whole brain lysates [115, 116].
Considering human risk variants, we have shown that microglia mediate the risk association between noncoding variation near LRRK2 and PD [45]. A noncoding variant, rs76904798, has the lowest p value for PD association at the LRRK2 locus on chromosome 12, with the T allele occurring more frequently in patients than controls [45]. Our lab used RNA sequencing of human donors and patient-derived iPSCs to demonstrate that individuals with one or more copies of the rs76904798 single nucleotide polymorphism (SNP) have higher LRRK2 expression and kinase activity in microglia than other cell types that might even have greater basal LRRK2 expression, identifying a microglia-specific eQTL [45]. Together, these studies demonstrate that microglial protein expression is subject to the influence of PD-relevant LRRK2 variation.
One immune-modulating pathway of interest in microglia is the TLR4 pathway. Activation of TLR4 can upregulate proinflammatory signaling by activating nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB), a transcription factor that promotes transcription of cytokines (ie. TNF-α, IL-1α), or via TIR-domain-containing adapter-inducing interferon-β (TRIFβ)-dependent downstream signaling that promotes transcription of interferons [117]. Lipopolysaccharide (LPS) is a protein found in the membrane of bacteria and a potent agonist of TLR4 [118]. LPS treatment leads to the phosphorylation and activation of LRRK2, leading to LRRK2-mediated phosphorylation of target Rab GTPases [40, 119–121]. LPS also triggers the upregulation of LRRK2 protein expression in microglia and peripheral immune cells in vitro and in the SNpc and striatum ex vivo, but it does not alter LRRK2 or TLR4 pathway mRNA expression [40, 122]. Furthermore, Gillardon et al. used immunoassays and differential expression analysis to demonstrate that the pathogenic LRRK2 p.R1441G mutation significantly increases LRRK2 protein expression and pro-inflammatory cytokine release from LPS-treated primary adult mouse microglial cells compared to WT controls [121]. Consistent with previous studies, they also showed that culturing primary cortical neurons in LPS-activated microglia conditioned media (MCM) increased neuronal death regardless of microglial genotype [121]. However, LPS-activated LRRK2 p.R1441G MCM led to much greater neuronal cell death than WT MCM [121].
Interestingly, a separate study showed that LPS can lead to elevated pro-apoptotic and pro-inflammatory signaling in the SNpc and striatum up to 2 months after treatment in LRRK2 p.R1441G mutant mice, while opposite effects have been demonstrated with LRRK2 loss of function [123]. Pretreatment of primary rat microglia with pharmacological LRRK2 kinase inhibitors have been shown to prevent microglia from undergoing morphological changes in response to LPS stimulation and to decrease TLR4-mediated proinflammatory cytokine release of TNF-α and its downstream targets, iNOS and phospho-p38 [40]. Similarly, microglia derived from LRRK2 KO rodents had a lower percentage of activated microglia and a reduced immune response to LPS treatment [124]. In addition, LRRK2 KO microglia have upregulated expression of CX3C motif chemokine receptor 1 (CX3CR1), a microglial receptor that suppresses inflammation and promotes motility, accompanied by increased microglial migratory distance and speed [124]. Double-knockout of LRRK2 and CX3CR1 restored microglial response to LPS, indicating that the attenuated immune response in LRRK2 KO cells is CX3CR1-dependent [124]. Overall, these studies suggest that the LRRK2 mutants promote microglial reactivity and a heightened immune response to inflammatory stimuli in a kinase-dependent manner while LRRK2 deficiency attenuates inflammatory responses. An obvious extension to this study would be to explore how cell type specific LRRK2 deficiency changes intercellular communication and cell fate in mixed culture models. For example, it would be interesting to co-culture LRRK2 knockout or wild type microglia with wild type neurons and determine how genotype modifies the effects of different immune response stimulants.
Another proinflammatory signaling pathway is the IFN-γ signaling pathway, which is involved in glial cell activation and innate and adaptive immunity [88]. IFN-γ is upregulated in the plasma, striatum, and SNpc of PD patients and IFN-γ stimulation increases LRRK2 expression in microglia, peripheral immune cells, and hiPSC-derived neurons [88, 125]. In contrast, treatment with LPS or IL-1 do not increase LRRK2 levels, demonstrating specificity of the effects of IFN-γ on LRRK2 expression [40]. Panagiotakopoulou et al. looked into the interactions between LRRK2 and IFN-γ and found that following IFN-γ stimulation, LRRK2 p.G2019S prevents the nuclear localization of NFAT, a key modulator of immune response, in hiPSC-derived microglia [88]. Furthermore, treatment of LRRK2 p.G2019S microglia with LPS or IFN-γ inhibits the nuclear translocation of NF-kB, also interfering with the TLR4 pathway [88]. Overall, pathogenic LRRK2 mutants with hyperactive kinase activity disrupt IFN-γ signaling and contribute to altered neuroimmune response in PD patients.
LRRK2 MUTATIONS CAN MODULATE MICROGLIAL MOTILITY AND PRUNING BEHAVIOR
Microglial behavior is also subject to the influence of PD-linked LRRK2 mutations. Microglia are highly motile cells because they must constantly survey the CNS microenvironment and quickly respond to any injury or pathogens [126]. When microglia sense purines released by damaged cells, they extend lamellipodia toward the site of damage [126]. Choi et al. conducted a study in which they found that primary microglia derived from LRRK2 p.G2019S transgenic mice had abnormal and diminished motility patterns compared to WT mice [126]. In response to adenosine diphosphate (ADP), WT microglia extended long protrusions and were highly mobile in the cellular environment for the duration of about 20 minutes, whereas LRRK2 p.G2019S microglia had limited movement and extended short protrusions that retracted quickly [126]. In contrast, LRRK2 kinase inhibition rescued G2019S microglial motility and LRRK2 knockdown BV2 microglia-like cells were faster, more mobile, and had denser actin structure than WT cells [126]. Corroborating this evidence, in vivo experiments showed that microglia in G2019S transgenic mice were slower to respond and less specific in isolating laser or stab-wound brain injury [126]. It was further suggested that LRRK2 directly interacts with focal adhesion kinase (FAK), a regulator of microglial migration, in the cytoplasm and mediates the inhibition of FAK activation via phosphorylation of the FAK-TXR (Thr–X–Arg/Lys) site [126]. LRRK2 p.G2019S mutant microglia and FAK-specific kinase inhibitors both decreased FAK activity and microglial movement in the presence of ADP stimulation, whereas LRRK2 knockdown and LRRK2 kinase dead mutants both increased FAK activation and movement [126].
During neural development, microglia prune excessive axons and regulate axonal outgrowth [127]. In the adult brain, microglia are still capable of synaptic pruning via phagocytosis [128]. One study found that in LRRK2 p.G2019S transgenic mice, microglia phagocytosed significantly more dopaminergic fibers at both 2 months and 10 months of age compared to WT mice [128]. Using confocal Z-stack imaging and 3D reconstruction, Zhang et al. quantified the size, number, and volume of dopamine transporter (DAT)+ or tyrosine hydroxylase (TH)+ puncta within ionized calcium binding adaptor molecule 1 (Iba1)+ microglia and found a higher number of TH+ and DAT+ structures and greater structure density in young and old LRRK2 p.G2019S mice compared to WT [128]. This study also explored microglial pruning activity in aged LRRK2 p.G2019S transgenic mice because these animals displayed mild hyperactivity despite declining motor coordination [128]. To identify the cause of the hyperactivity, the authors then investigated excitatory synapse density in the prefrontal cortex and striatum [128]. In aged LRRK2 p.G2019S mice, microglia had significantly fewer postsynaptic density 95 (PSD95)- and vesicular glutamate 1 (VGLUT1)-positive intracellular puncta [128]. In contrast, there were significantly higher extracellular levels of PSD95 and VGLUT1 and greater dendritic spine density in the PFC, suggesting dysregulated microglial phagocytosis of excitatory synapses [128]. Collectively, these studies show that pathogenic LRRK2 mutations can disrupt microglial motility and phagocytosis, potentially altering intercellular communication and downstream cellular response.
LRRK2 MUTATIONS CAN INDIRECTLY CONTRIBUTE TO OXIDATIVE METABOLISM
Within microglia, LRRK2 regulates two processes critical to healthy oxidative metabolism, namely mitochondrial fission dynamics and iron homeostasis [64, 130]. Mitochondria have been shown to influence ROS production as well as NF-kB-mediated and MAPK-mediated proinflammatory cytokine release in activated microglia [131]. Ho et al. published two studies exploring the impact of LRRK2 on mitochondrial fission in primary microglia and BV2 microglia-like cells [115, 116]. Based on image analysis and increased expression of the mitochondrial fission biomarker dynamin-related protein 1 (Drp1), LPS treatment increases mitochondrial fission, but this effect is abolished when LRRK2 kinase activity is inhibited by GSK2578215A [115]. Ho et al. also used whole brain lysates and primary microglia derived from transgenic LRRK2 p.G2019S and WT mice to show that the G2019S mutation increases mitochondrial fission, and this effect is upregulated with age [115]. Confirming this effect, BV2 cells transfected with myc-tagged LRRK2 p.G2019S had decreased mitochondrial perimeter, increased quantity of mitochondria per cell, and increased Drp1 expression, indicating significantly higher levels of mitochondrial fission [116].
Dysregulation of iron homeostasis is one of the many processes thought to underlie neurodegeneration and iron deposition in the brain is seen in PD patients [130]. Iron is crucial for oxidative metabolism, mitochondrial energy production, and synthesis of neurotransmitters [130]. However, excess iron can lead to metabolic changes such as decreased respiration and increased ROS production [130]. Chronic neuroinflammation leads to intracellular sequestration and accumulation of iron and activated microglia uptake more iron from the extracellular environment [130]. The primary method of iron transport into cells is uptake mediated by transferrin, an iron carrier protein [130]. However, microglia polarization alters uptake of iron such that microglia preferentially uptake transferrin (Tf)-bound iron in the presence of anti-inflammatory stimuli and non-Tf-bound iron in the presence of pro-inflammatory stimuli [129]. After iron binds Tf, the complex binds to Tf receptors and gets internalized by the cell via endocytosis and then either rapidly recycled back to the plasma membrane or slowly recycled via Rab8-positive endocytic recycling compartments (ERCs) [129]. Previous research from our group explored the effects of pathogenic LRRK2 mutations on iron uptake and Tf recycling in the context of neuroinflammation [129]. In HEK293T cells, expression of pathogenic LRRK2 mutations (p.R1441C, p.Y1699C, p.G2019S, or p.I2020T) led to the sequestration of phosphorylated Rab8 at lysosomes and in turn, mistrafficking of Tf receptors to lysosomes instead of the endocytic recycling compartment [129]. Mass spectrometry then revealed elevated iron levels in G2019S cells compared to WT [129]. RNAseq analyses also showed that genes involved in the endolysosomal system, vesicular trafficking, and iron homeostasis were differentially expressed in microglia under inflammatory conditions in vivo and in vitro [129]. In addition, p.G2019S knock-in mice had significantly increased iron accumulation and levels of the iron storage protein ferritin in striatal microglia following LPS injection [129]. Thus, mutations in LRRK2 may cause iron dysregulation in microglia in the context of the chronic neuroinflammation seen in PD.
LRRK2 MUTATIONS CAN INFLUENCE PERIPHERAL IMMUNE CELL HOMEOSTASIS AND SIGNALING
LRRK2 is also highly expressed in immune cells outside of the CNS including monocytes, macrophages, dendritic cells, and neutrophils [132]. In peripheral immune cells, infection by intracellular pathogens such as Mycobacterium Tuberculosis has been shown to stimulate LRRK2 kinase activity [133]. In a 2020 study, Herbst et al. demonstrated that phagosome membrane damage is one mechanism by which pathogens recruit and activate LRRK2 at phagosomal membranes in peripheral immune cells, and furthermore, go on to establish a possible role for LRRK2 in membrane repair [133]. In a separate study, Shutinoski et al. showed that LRRK2 p.G2019S transgenic mice had LRRK2 kinase-dependent reduced bacterial load, improved antimicrobial immune response, and prolonged survival to sepsis when acutely infected with gram-positive Salmonella Typhimurium [134]. In contrast, LRRK2 p.R1441C transgenic mice did not have differences in bacterial load compared to wildtype animals [134]. The same study also showed that LRRK2 p.G2019S transgenic mice had lower viral burden and increased leukocyte recruitment when given a brain infection with reovirus-T3D [134]. Considering the role of LRRK2 in peripheral pathogen response, future studies should examine the role of microglial LRRK2 in response to bacterial and viral pathogens in the brain.
Overall, these results suggest that LRRK2 has a generally pro-inflammatory effects on microglia. Mechanistically, LRRK2 likely affects inflammatory processes via effects on the endo-lysosome system. Importantly, the available data suggest that lysosomal function and inflammation are interrelated with each other. It is likely that LRRK2 plays a role in lysosomal damage response, again in the context of inflammatory signaling.
CONCLUSION
The literature summarized in this review highlights the importance of LRRK2 in intracellular signaling in neurons, astrocytes, and microglia. The research reviewed strongly demonstrates that PD-causing mutation, LRRK2 p.G2019S and p.R1441C, disrupt cellular homeostasis and function. However, it remains unclear as to how LRRK2 mutations lead to PD pathogenesis.
It can be concluded from this review that LRRK2 is a key mediator of endolysosomal function in neurons, astrocytes, and microglia. PD-causing LRRK2 mutations disrupt endolysosomal function such that it interferes with downstream gene expression, intercellular communication, and degradation of neurotoxic protein aggregation. Particularly of interest for the PD field may be the indirect effects of LRRK2-mediated lysosomal disruption on Lewy body accumulation. However, the specific lysosomal alterations that occur in the presence of PD-linked LRRK2 mutations remain unresolved and must be further explored to resolve the link between LRRK2, lysosomal function, and protein aggregation. As discussed above, neurons cultured from LRRK2 p.G2019S knock-in mice had significantly smaller lysosomes than wildtype murine neurons, whereas astrocytes overexpressing LRRK2 p.G2019S had enlarged lysosomes. Along the same lines, is that the literature reveals conflicting results on pH changes in the presence of PD-linked LRRK2 mutations. One central argument that we have tried to advance here is that comparison between studies performed in different models, including different mutations and model using overexpression or modification of endogenous LRRK2, is made difficult by each study focusing on one cell type. The field would benefit from future studies comparing the effect of PD-linked LRRK2 mutations on multiple CNS cell types derived from the same model (i.e. patient tissue, mice with the same genetic manipulation, etc.) that utilize a standardized set of assays to measure endolysosomal changes.
It is also important to note that many of the studies reviewed in this article utilize monoculture models and therefore, do not address potential non-cell-autonomous effects that would be present in a physiological context. While exploring LRRK2 biology and physiology within individual cell types in the CNS is an important first step, it will be crucial for future research to expand upon the indirect effects of LRRK2 mutations in glial cells on neuron health and survival to understand how LRRK2 may facilitate PD pathogenesis. Considering the recent studies done showing that LRRK2 mutations in glial cells can lead to neuronal dysfunction and protein accumulation, we hypothesize that altered intercellular interactions likely drive PD pathogenesis. Future studies should consider dual and triculture models to improve physiological relevance of LRRK2 research. In addition, the genetic manipulation of LRRK2 in vivo may provide a better understanding of how LRRK2 can impact intercellular interaction.
Finally, this review also shows the emphasis of current research on the LRRK2 p.G2019S mutation and a relative lesser investigation into the effects of other PD-causing LRRK2 mutations on CNS cell types, exposing a critical area for future research. It would also critical to consider multiple mutations that, in humans, all converge on PD as a phenotype as it is likely that shared rather than distinct consequences of mutations are critical for PD pathogenesis.
Footnotes
ACKNOWLEDGMENTS
The authors have no acknowledgments to report.
FUNDING
This research was supported entirely by the Intramural research Program of the NIH, National institute on Aging.
CONFLICT OF INTEREST
Mark R. Cookson is an Editorial Board Member of this journal, but was not involved in the peer-review process nor had access to any information regarding its peer-review.