
Abstract
The increasing global burden of Parkinson’s disease (PD), termed the PD pandemic, is exceeding expectations related purely to population aging and is likely driven in part by lifestyle changes and environmental factors. Pesticides are well recognized risk factors for PD, supported by both epidemiological and experimental evidence, with multiple detrimental effects beyond dopaminergic neuron damage alone. The microbiome-gut-brain axis has gained much attention in recent years and is considered to be a significant contributor and driver of PD pathogenesis. In this narrative review, we first focus on how both pesticides and the microbiome may influence PD initiation and progression independently, describing pesticide-related central and peripheral neurotoxicity and microbiome-related local and systemic effects due to dysbiosis and microbial metabolites. We then depict the bidirectional interplay between pesticides and the microbiome in the context of PD, synthesizing current knowledge about pesticide-induced dysbiosis, microbiome-mediated alterations in pesticide availability, metabolism and toxicity, and complex systemic pesticide-microbiome-host interactions related to inflammatory and metabolic pathways, insulin resistance and other mechanisms. An overview of the unknowns follows, and the role of pesticide-microbiome interactions in the proposed body-/brain-first phenotypes of PD, the complexity of environmental exposures and gene-environment interactions is discussed. The final part deals with possible further steps for translation, consisting of recommendations on future pesticide use and research as well as an outline of promising preventive/therapeutic approaches targeted on strengthening or restoring a healthy gut microbiome, closing with a summary of current gaps and future perspectives in the field.
Keywords
INTRODUCTION –THE INCREASING GLOBAL BURDEN OF PARKINSON’S DISEASE AND POSSIBLE LINKS TO ENVIRONMENTAL EXPOSURES
Parkinson’s disease (PD) is the second-most common neurodegenerative disorder, affecting 2–3% of the population≥65 years of age. While we still lack a comprehensive understanding of the etiology, the underlying α-synuclein (α-syn) pathology and (neuro)inflammation are widely recognized as key players driving PD progression [1], although other factors, such as tau pathology, are likely important to a lesser extent [2, 3]. Among neurological disorders, PD has undergone the fastest growth in prevalence and disability in recent years and has become one of the leading causes of disability worldwide [4, 5]. According to data obtained from the Global Burden of Disease 2019, increasing trends of PD burden were observed globally in most regions and countries [6]. This allows us to talk about a Parkinson’s pandemic [7] similar to the pandemic of other non-communicable diseases—fueled by social, political, economic and environmental trends and changes [8]. The aging of the population contributed substantially to the rise in the global prevalent number of PD cases from 1990 (2.5 million) to 2019 (8.5 million), but an upward trend is also evident in the age-standardized rate of prevalence and incidence, indicating that global aging may not be the only reason for the increased prevalence, and other factors are likely important [6]. Although socioeconomic level is, in general, positively associated with health [9], the opposite seems to be true for PD, with age-standardized disability-adjusted life-years due to PD being increased with higher sociodemographic index (SDI) [5]. This, on the one hand, may be related to longer survival of more wealthy PD patients due to more sophisticated and also more expensive treatment options. On the other hand, growing world industrialization, pronounced in countries with higher SDI, may be linked to several environmental factors driving the increased global burden of PD, such as exposure to pesticides [10], solvents [10] or metals [11, 12]. The largest increase in the age-adjusted prevalence rate of PD between 1990 and 2016 worldwide was documented in China, a middle SDI country that has undergone rapid industrial growth since 1990 [5]. Occupational exposure and industrial pollution also accelerated the increasing trends in low and middle SDI areas [6, 14]. These factors are particularly interesting, as they represent a major challenge to public health as well as an open door for risk modification on a large scale.
The association of pesticides exposure and risk of PD is well known [10, 16], being included as an independent risk factor for PD in the original and updated version of the International Parkinson and Movement Disorders Society (MDS) research criteria for prodromal PD (positive likelihood ratio of 1.5) [17, 18]. Based on several meta-analyses [10, 20] and expert reviews, PD was recognized as an occupational disease in agriculture professionals in France in 2012 (under the conditions of a diagnosis confirmed by a neurologist and professional exposure to pesticides for 10 years or more) [21]. The epidemiological evidence for a link between PD and pesticides is strong, yet the global use of pesticides is still at or near its highest levels, including those with a clear impact on PD pathogenesis, such as paraquat. Some measures have been taken, and paraquat has been gradually banned in almost 60 countries, including Switzerland (1989), the United Kingdom, the European Union (2007) [22], South Korea (2011) [23], China (2016) [24] or lately Brazil and Taiwan (2020). However, the export to other countries, including African countries, India, Australia, or the United States, continues [7].
Despite the already established connection between pesticides and PD, the exact mechanisms of how pesticides contribute to PD pathology are still under investigation. While direct effects of pesticides on the central nervous system (CNS) have been extensively investigated, especially in the mouse model of PD, effects on the peripheral (PNS) and the enteric nervous system (ENS) are not fully understood yet. The microbiome-gut-brain axis, the bidirectional communication between the ENS and the CNS, including the vagus nerve, endocrine as well as immune pathways and bacterial metabolites, has been in the scope of PD research in recent years [25–27]. In this regard major focus has been placed on the influence of nutrition, pre- and probiotics or fecal microbiota transplantation (FMT) on the microbiome with regard to possible future treatment strategies [28]. However, the impact of pesticides on the microbiota has started to gain more attention only recently, since they may have substantial detrimental effects on microbiome composition and function, which may play an important role in the pathophysiology of PD and other neurodegenerative and non-communicable diseases [29–32].
We acknowledge the tremendous work already done in this area of research, as represented by multiple reviews targeting the topics of environmental exposures and risk of PD, as well as the role of the microbiome-gut-brain axis in the pathogenesis of PD independently; emerging attention is starting to be paid to the impact of pesticides on the microbiome in general. This review aims to integrate these separate perspectives together in the context of PD development, summarize the direct effects of PD-relevant pesticides on the CNS and the ENS and investigate their impact on the microbiome and microbiome-gut-brain axis, represented by both local and systemic effects of dysbiosis, including leaky gut with easier penetration of toxins through the intestinal barrier and multiple subsequent cascades mediated by bacterial metabolites. To remain practically and clinically oriented, an overview of possible future steps for translation follows, consisting of recommendations for future pesticide use and research.
METHODS
A search in Medline (Pubmed) was conducted using terms referring to Parkinson’s disease, pesticides and microbiome, as follows: (“Parkinson Disease” [Mesh] OR “Parkinson*” [tw]) AND (“Pesticides” [Mesh] OR “Agrochemicals” [Mesh] OR “Pesticide Residues” [Mesh] OR “Pesticide Synergists” [Mesh] OR “pesticide*” [tw]) AND (“Microbiota” [Mesh] OR “Brain-Gut Axis” [Mesh] OR “Microbiotas” [tw] OR “Microbial Community” [tw] OR “Community, Microbial” [tw] OR “Microbial Communities” [tw] OR “Microbial Community Composition” [tw] OR “Community Composition, Microbial” [tw] OR “Composition, Microbial Community” [tw] OR “Microbial Community Compositions” [tw] OR “Microbial Community Structure” [tw] OR “Community Structure, Microbial” [tw] OR “Microbial Community Structures” [tw] OR “Human Microbiome” [tw] OR “Human Microbiomes” [tw] OR “Microbiome, Human” [tw] OR “Microbiome” [tw] OR “Microbiomes” [tw] OR “gut-brain axis” [tw] OR “brain-gut axis” [tw]).
Additional relevant articles were detected in the citation lists of the papers identified by the literature search. All types of articles published in English up until December 2022 were evaluated for this narrative review.
DIRECT EFFECTS OF PESTICIDES ON THE NERVOUS SYSTEM
The recognition of a potential link between pesticides and PD dates back to the 1980s, when several individuals developed marked parkinsonism after intravenous illicit drug abuse. Exposure to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), a substance structurally similar to the herbicide paraquat, led to selective damage of dopaminergic (DA) neurons in the substantia nigra (SN) [33]. Since then, several pesticides have been used in experimental and animal (mostly rodent) models of PD [34]: MPTP (selective death of DA neurons in the SN) [35], rotenone (inhibition of complex I in the respiratory chain, selective nigrostriatal DA degeneration, cytoplasmic inclusions similar to Lewy bodies, and motor symptoms) [36, 37], paraquat (dose-dependent degeneration of DA neurons in the SN) [38, 39], the combination of paraquat and maneb (degeneration of DA neurons in the SN, loss of striatal dopamine and reduced motor activity) [40], or dieldrin (increased α-syn expression and alterations in the DA system) [41]. Pesticide-induced PD animal models serve as a proof-of-principle concept that exogenous substances can produce selective degeneration of DA neurons in the SN, i.e., that environmental toxicants can have a direct role in development of PD [42]. Since the 1980s, multiple studies have unraveled the connection between pesticides and PD from both epidemiological [43, 44] and molecular perspectives; these were recently reviewed, e.g., by Yuan et al. [45], Vellingiri et al. [46], or Nabi et al. [47]. Apart from MPTP, the pesticides most implicated in PD include rotenone, paraquat, maneb, organochlorines, and pyrethroids. All of them are still widely used in agriculture or aquaculture as herbicides, fungicides, or insecticides [29]. Abundant application leaves residues or metabolites of pesticides in food, drinking or groundwater [48], indicating different possible effective entry routes for humans, though in most cases, it is the gut that is directly in contact with food contaminants [29]. However, in cases of occupational contact with pesticides, exposure via inhalation and ingestion has also been associated with a higher risk of late-life prodromal features of PD than dermal exposure only [49].
A clear distinction between toxin-induced parkinsonism and idiopathic PD may be complicated. Cases with acute-onset tremors, bradykinesia and rigidity following accidental or voluntary exposure, usually not progressing once the agent is stopped, with 10% to 20% even improving (described, e.g., in manganese, mercury, MPTP, organochlorines, organophosphates, rotenone, or paraquat exposure [50, 51]) clearly fall into the category of toxin-induced parkinsonism. However, it may not be possible to draw this dividing line so unequivocally in the case of epidemiological studies linking higher PD prevalence with certain geographic regions or occupations. A comparison to the genetic architecture of PD can be made—there is a genetic background continuum ranging from rare high penetrance gain or loss of function mutations (causing PD on their own, often with a clinical phenotype different from idiopathic PD [52]) to relatively common genetic risk variants that only mildly modify disease risk [53]. The same may be true for pesticides, depending on the intensity and duration of exposure. Cases of toxin-induced parkinsonism following acute high-dose accidental or intentional pesticide intoxication represent their direct causative effect [33, 55], whereas increased PD prevalence among agricultural workers with chronic occupational exposure or in areas of chronic residential exposure [56] might represent their contribution to disease pathogenesis as a modifier of disease risk (although other factors are likely important, as not all equally exposed workers subsequently develop PD; pesticides might act as one piece of the mosaic in the multifactorial etiology of PD) [21, 57].
A recent quantitative epidemiological study aimed to identify PD-relevant pesticides; out of 288 specific pesticides available for PD risk evaluation in a pesticide-wide association study, long-term exposure to 53 of them was associated with PD, with 10 pesticides being directly toxic to DA neurons derived from PD patient-induced pluripotent stem cells. Co-exposures resulted in greater toxicity than any single pesticide, and trifluralin was found to be a driver of toxicity to DA neurons, leading to mitochondrial dysfunction [58]. These findings bring new pollutants into research attention and pave the way forward for understanding the effects of complex pesticidal exposure. However, as the evidence on their molecular mechanisms and impact on gut microbiota is currently missing, the focus of this review will be on pesticides with a long-recognized link to PD and their impact on disease pathogenesis through multiple pathways.
Central effects: toxicity on dopaminergic neurons in the substantia nigra
Pesticides associated with PD lead to α-syn pathology and degeneration of DA neurons through a number of molecular mechanisms, including oxidative stress, interference with dopamine transporters (DAT), mitochondrial abnormalities, stimulation of α-syn fibrillation and neuroinflammation [46]. The most relevant pesticides and their effects are mentioned in the following text.
Taken together, there is very strong evidence from rodent models linking pesticides to dopaminergic toxicity, particularly for rotenone and paraquat.
Peripheral effects: toxicity on the gut and enteric nervous system
Gut dysfunction and the gut-brain axis are of particular interest due to their possible involvement in the pathogenesis of PD [104]. In terms of the discovered nigro-vagal pathway [105], ingested environmental toxicants could enter the CNS through vagal connections from the gut and induce or accelerate PD progression [104]. In fact, beyond pure DA neuronal damage in the SN, peripheral toxicity, and alterations of the ENS/PNS were described after exposure to several pesticides, as outlined in the following text.
In a
In
In models studying
The extent of current knowledge about the impact of various pesticides on the ENS/PNS in PD is still rather limited compared to the level of evidence of their central neurotoxicity to DA neurons in the SN. However, this research area may be of particular interest in the context of increasing knowledge on the role of the microbiome-gut-brain axis in PD development, as discussed in the next section.
PESTICIDES AND THE MICROBIOME-GUT-BRAIN AXIS
The microbiome-gut-brain axis in PD
The prodromal phase of PD
To elucidate potential options about how pesticides might influence PD pathology and progression, it is important to outline how PD pathology progresses throughout the nervous system before a diagnosis can be made based on the onset of cardinal motor symptoms. It is now accepted that PD has a long prodromal phase in which pathology spreads throughout the peripheral and central nervous system before a wide-range pathology in the SN results in the onset of cardinal motor symptoms [122]. The “dual hit hypothesis”, proposed in 2007, suggested a nasal route (with olfactory bulb involvement and anterograde progression to the temporal lobe) versus a gastrointestinal route of entry (with propagation of pathology via the vagus nerve) [123]. In fact, both hyposmia and gut dysfunction are common non-motor symptoms not only in manifest PD, but also preceding the onset of motor symptoms by years [124]. The gut has become particularly interesting as an early disease indicator, with constipation being the earliest non-motor symptom documented (up to 20 years prior to onset of motor symptoms, even before hyposmia) [125]. Indeed, seeding the duodenum with α-syn preformed fibrils in a mouse model led to disruption of ENS connectivity and time-dependent GI dysfunction [126]. In humans, α-syn deposits in GI tissues have been observed up to 20 years prior to a PD diagnosis [127], although we still lack full understanding of the diversity of α-syn misfolded forms, which does not allow its use as a reliable PD biomarker yet, despite significant efforts to develop a standardized diagnostic tool [128, 129]. Thus, at least in a subtype of PD patients the gastrointestinal tract (GIT) may be the starting point where neuroinflammation and neurodegeneration leading to PD pathology might be triggered. Environmental pathogens might then gain access through the GIT, disrupting the ENS and leading to α-syn pathology, which could reach the brain in a prion-like manner via the vagus nerve [130, 131].
Two basic different PD subtypes have been proposed in regards to the trajectory of disease progression: the so-called brain-first and body-first PD [132]. In the proposed brain-first PD the initial α-syn misfolding happens in the CNS with subsequent descendent spreading [133]. In contrast, the body-first subtype is characterized by initiation of α-syn misfolding in the gut (ENS/PNS) and ascendent α-syn pathology through the vagus nerve to the brainstem and higher [122], and changes in the microbiome-gut-brain axis induced by pesticide exposure are likely more relevant for this phenotype, as discussed in the following text.
Enteral dysbiosis in PD
Gut health is a precondition for brain health; alterations of the gut microbiota composition are observed to co-occur with many neurological diseases [134]. The gut microbiome, composed of thousands of diverse microbial species, is involved in multiple complex interactions with the human body, including metabolism (synthesis and degradation of vitamins, amino acids, lipids, bile acids (BA) and neurotransmitters; fermentation of indigestible food substances, xenobiotic metabolism), immune function (stimulation and/or regulation of immune responses) or structural function (regulation of epithelial cell growth and the intestinal barrier) [135]. Host-microbiome crosstalk has come into focus as the key mediator of neurological health; there is evidence that gut microbiota regulate the development of the blood-brain-barrier (BBB) and influence the function of microglia and astrocytes [136]. Recent reviews have summarized the role of gut microbiota in risk of developing neurodegenerative diseases [137, 138] and PD in particular [139, 140], even focusing on the prodromal period [141]. Taken together, a number of studies have detected alterations in gut microbiota composition in PD patients, with subsequent changes in their products and metabolites. In the summary of Tan et al., at least 42 families, 102 genera and 44 species of bacteria were reported to be differentially abundant in PD patients compared to controls [139]. From a functional point of view, this dysbiosis might result in a pro-inflammatory status which could be linked to the recurrent GI symptoms affecting PD patients.
Local and systemic effects of dysbiosis in PD: “leaky gut”, inflammation, and α-syn pathology
Intestinal integrity is crucial for proper gut function. The semipermeable gut barrier limits the transport of harmful substances (including environmental pollutants), regulates absorption of nutrients and allows immune sensing, whereas a “leaky” gut, a condition of compromised intestinal barrier seen in PD, results in an increased flux of both microbes (including bacteria) and molecules (including toxins) across the intestinal epithelium. Various studies support the concept of intestinal hyperpermeability and inflammation in PD, including among others increased fecal markers of intestinal inflammation [142, 143] and higher pro-inflammatory gene profiles associated with intestinal barrier disruption [144]. Dysbiotic gut microbiota expressing more lipopolysaccharides (LPS) can overstimulate the toll-like receptors (TLR) expressed on epithelial, immune and enteric glial cells, which together with higher intestinal barrier permeability further provokes local and systemic inflammation and enteric neuroglial activation, triggering the development of α-syn pathology and ultimately not only gut, but also brain inflammation [113, 145–150].
It has been suggested that α-syn participates in innate and adaptive immunity [151, 152], and inflammation is connected to an important feedback loop that promotes its spread –an inflammatory environment enhances α-syn expression, misfolding and aggregation [148, 153–158], which in turn induces local proinflammatory immune responses [126, 160]. Gut microbes, besides contributing to inflammation, can play an additional mechanistic role in the process, as microbial curli (extracellular amyloid proteins produced and expressed abundantly by certain bacteria, such as Escherichia coli) have been suggested to template α-syn aggregation through cross-seeding mechanism in the gut [161, 162].
Tan et al. summarized the whole process as a vicious cycle where dysbiosis, hyperpermeability, inflammation, and α-syn aggregation drive and perpetuate one another [139]. Notably, as gut inflammation and expression of α-syn might be common events during life, other additional contributing factors are proposed as having importance for driving PD pathogenesis in vulnerable persons, including environmental influences [163, 164].
According to the current understanding, there is a process that begins locally—leaky gut and dysbiosis-induced gut inflammation (detected in both colonic biopsies and fecal samples from PD patients [142–144, 148–150]) which then continues to drive disease pathogenesis through several systemic mechanisms, such as increased cytokine production, BBB disruption, migration of inflammatory cells into the brain [160] and microglial activation, finally culminating in neuronal dysfunction and/or loss [165].
Pesticides affect the microbiome: consequences of pesticide-induced dysbiosis
The impact of environmental pollutants (and pesticides in particular) on gut health has started to gain recognition in recent years [30]. There is sufficient evidence that pesticides can impair gut barrier function by destroying intestinal mucosa and affecting the crosstalk between mucus and gut bacteria [166, 167]. They alter the overall composition of gut microbiota and affect their metabolites, leading to pathological changes by acting on receptor sites of different tissues and organs [168]. It is time to seriously consider the gut microbiota as an unintended recipient of pesticidal pollution and to focus on the long-term effects of chronic exposure on microbial diversity and subsequent influence on the host [29]. Table 1 shows a summary of the shifts in gut microbial composition following exposure to several PD-relevant pesticides in various experimental animal models, which can also be found in the following section.
Shifts in the gut microbial composition following exposure to PD-relevant pesticides in various experimental models
Rotenone
In a rotenone-induced mouse model of PD, a clear shift towards putative pro-inflammatory dysbiosis was observed in both the cecal mucosa-associated and luminal microbiota community structure. Changes in β-diversity were represented by a decreased Firmicutes-to-Bacteroidetes (F/B) ratio, increased relative abundance of putative pro-inflammatory bacteria, such as Rikenellaceae and Allobaculum, and decreased relative abundance of putative anti-inflammatory bacteria, such as Bifidobacterium. Dysbiotic microbiota correlated with PD-like functional and pathological changes in both the gut and the brain, as seen in the association of above-mentioned bacterial families with intestinal epithelial barrier integrity (assessed by ZO-1 expression), inflammation (assessed by the number of CD3 + T-cells in the colon), α-syn accumulation in the colonic plexi and the number of DA neurons in the SN. Moreover, rotenone induced a significant reduction of several metabolic pathways in the gut microbiota: biodegradation and metabolism of xenobiotics, as well as metabolism of cofactors, vitamins, carbohydrates, amino- and fatty acids. Another study in a mouse model observed rotenone-induced GI and motor dysfunctions that correlated with changes in fecal microbiota composition; dysbiosis was characterized by an overall decrease in bacterial diversity and a significant microbial shift in the main bacterial phyla, resulting in an increased F/B ratio [170], the last being in contrast with the previous study. The differences were explained by possible effects of different rotenone oral dose concentrations, different GI sites and different sequencing techniques [169]. Another study found an increase in the relative abundance of species of the genera Lactobacillus, Bifidobacterium, Akkermansia, and Bacteroides and a decrease in species of Lachnospiraceae, Ruminococcaceae, Turicibacter, Faecalibaculum, and Clostridium in rotenone-treated mice [171]. Several of these shifts (increased Lactobacillaceae and Bacteroides and decreased Lachnospiraceae) were consistent with findings from human PD studies, where they were associated with worsening of clinical symptoms, such as gait disturbances or postural instability [172].
In a rat model, intraperitoneal rotenone administration led to alterations of small intestinal and colonic microbiome composition (although most of them failed to reach statistical significance, with the exception of increased Bifidobacterium in the colon) and reproduced clinical symptoms of gastroparesis before nigrostriatal pathology was evident [173]. Several microbial shifts were in line with changes reported in human PD patients: increased Lactobacillus and Bifidobacterium [174–177] and increased Clostridiaceae and reduced Lachnospiraceae and Prevotellaceae [175, 179]. Decreases in the Prevotellaceae family and increases in the Lactobacillaceae and Bifidobacteriaceae families are of particular interest, as these changes have been associated with reduced serum ghrelin [177, 180], a gut hormone that reportedly regulates nigrostriatal dopamine function and thus may be able to attenuate neurodegeneration in PD [181]. Thus, animal rotenone PD models seem to replicate human PD-related dysbiosis, with subsequent inflammatory changes in the gut and an influence on further metabolic pathways.
Paraquat
Intraperitoneal paraquat administration in a mouse model of PD led to alterations in gut microbiome diversity: the relative abundance was significantly different in 10 dominant bacterial groups, with Lachnospiraceae, Ruminococcaceae, and Rikenellaceae being the lowest and Bacteroides the highest after four weeks, while the relative abundance of Prevotellaceae was significantly lower already after two weeks. Besides gut microbiota disruption, intestinal epithelial barrier damage (seen as decreased expression levels of colonic tight junction proteins) and inflammatory responses (seen as increased expression levels of inflammatory markers) were suggested to be the main causes of gut dysfunction, and pathological aggregation of gut-derived α-syn and disturbance of short-chain fatty acids (SCFA) in the gut might be the key components in communication of the gut-brain axis, leading to pathological changes in the CNS after paraquat exposure [182]. Interesting results were observed in another study: postnatal paraquat intraperitoneal injection in mice increased the body weight and reduced the gut microbiota diversity of adult mice males, with gene function prediction analysis suggesting that the two observed outcomes were highly correlated [183]. Although this was not a mouse model specific for PD, it demonstrates that early-life paraquat exposure can disturb the gut microbiota and result in long-term effects in a sex-specific manner, and given that obesity, metabolic syndrome and type 2 diabetes are risk factors for PD [184] and that PD more frequently affects men, these findings might also be relevant.
Glyphosate
Mode of action, alterations in the gut microbiota composition: Several effects of glyphosate (Gly) are proposed to be mediated by inducing alterations in gut microbiome composition. Gly (herbicide) acts as a competitive inhibitor of 5-enolpyruvylshikimate-3-phosphate (EPSP) synthase, an enzyme involved in the shikimate pathway responsible for the synthesis of aromatic amino acids (phenylalanine –Phe, tyrosine –Tyr and tryptophan –Trp) in plants, essential for cellular survival [185]. Gly was previously considered to be a safe compound for humans, as EPSP synthase is absent in animals and humans, who depend upon ingested food and gut microbes to provide these essential nutrients [120]. However, the shikimate pathway is essential to the metabolism of some species in the human gut microbiome. A conservative estimate is that 54% of species in the core human gut microbiome are sensitive to Gly [186]. Thus, Gly exposure leads to enzymatic inhibition in bacteria and subsequent gut dysbiosis, which causes behavioral impairments [187].
A number of studies detected various gut microbiome imbalances in animal models, although not specifically related to PD. In mice, a significant alteration in gut microbiota composition, abundance and phylogenetic diversity was observed for Corynebacterium, Firmicutes, Bacteroides and Lactobacillus, all reduced by Gly exposure; furthermore, increased anxiety and depression-like behavior occurred after subchronic and chronic exposure to GBHs herbicides [188]. In a rat model, dysbiosis induced by Gly was characterized by an increase in Bacteroidetes over Lactobacillus, with a sex-specific effect, principally affecting female rats [189]. Similar results were also seen in rat pups after maternal exposure to GBHs in drinking water. The relative abundance of Bacteroidetes (Prevotella) was increased, and Firmicutes (Lactobacillus) was reduced [190]. A reduction of the bacterial community and a weakening of beneficial bacterial composition was observed in honey bees, which makes them more susceptible to infection by Serratia, an opportunistic pathogen causing mortality [191]. In poultry microbiota, a variable effect on different bacterial strains was documented: beneficial intestinal bacteria, such as Enterococcus faecalis, E. faecium, Bacillus badius and B. cereus, were found to be sensitive to Gly, whereas the Clostridia species and pathogenic bacteria, such as E.coli, Salmonella enteritidis, S. typhimurium or S. galliarum, showed marked resistance to Gly [192]. Varying results were found in cow rumen: a reduction in Entodinium, Epidinium, Ophryoscolex, and Dasytricha species [193] compared to no changes in another study [194]. In horse and cattle, Gly caused low levels of Enterococcus species, which altered its inhibitory role against Clostridium botulinum [195]. A predisposition to the development of Clostridium tertium bacteraemia in humans with an effect on the intestinal mucosa was indicated after a voluntary ingestion of Gly (a suicide attempt by a human female) [196]. Consequently, it has been suggested that Gly can cause dysbiosis [120]. However, relevance for the human gut microbiome remains questionable. Most gut bacteria do not possess a complete shikimate pathway; this pathway is mostly transcriptionally inactive, suggesting that gut bacteria are mostly aromatic amino acid auxotrophs (in conditions of adequate dietary quantity) and are thus relatively resistant to potential growth inhibition induced by Gly. Moreover, one-fifth of E. coli EPSPS enzyme homologues have been found to be resistant to Gly, and antimicrobial effects were mostly found at high doses unlikely to occur in real-world exposure scenarios [197].
Alterations in neurotransmitter metabolism: There are several mechanisms that are unique to Gly among all emergent herbicides. Inhibition of the shikimate pathway not only has deleterious effects on gut microbiota metabolism and survival itself, but also leads to reduced synthesis of essential aromatic amino acids humans depend on. Apart from Trp, Tyr and Phe, there are other biologically active molecules that need shikimate pathway metabolites as precursors (serotonin, melatonin, melanin, epinephrine, dopamine, thyroid hormone, folate, coenzyme Q10, vitamin K, vitamin E); methionine and glycine are also negatively impacted by Gly [198].
Gut bacteria interact with the host nervous system through several neurotransmitters or their metabolic precursors [199], including GABA and Trp, the latter of which is a central precursor of serotonin (5-hydroxytryptamine, 5-HT), a neurotransmitter involved in many different physiological processes and psychoaffective functions [200]. The gut microbiota has already been repeatedly shown to participate in modulation of 5-HT turnover [201]. Trp is generated by the gut microbiota and is converted to 5-HT after crossing the BBB [202]. Returning to the shikimate pathway again, Gly acts as an inhibitor of EPSP synthase, the rate-limiting step in the synthesis of aromatic amino acids, including Trp [198]. Indeed, Gly effects in altering gut microbiota composition with subsequent neurobehavioral consequences were implicated in several conditions, such as depression and anxiety [188] or autism spectrum disorders [203], reviewed in [120], even in case of maternal exposure with outcomes in the offspring, suggesting that microbiota could regulate neurobehavior by modulating the development of CNS serotonergic neurotransmission [188]. Given that PD is a multi-transmitter neurodegenerative disorder with a significant role of serotonergic signaling in many non-motor symptoms, mainly neuropsychiatric [204, 205], this mode of action of Gly might also be of potential relevance in PD.
Manganese metabolism interference: Manganese (Mn) seems to be another piece of the mosaic regarding complex PD risk from environmental exposure. Manganism, a condition closely resembling PD (with similar clinical manifestation, although some differences in underlying neuropathology—the absence of Lewy bodies or different distribution of neuronal loss—in the locus coeruleus, globus pallidus and pars reticulata of the SN [206, 207]) develops after chronic occupational Mn exposure [208, 209], and there is an increase in the incidence of PD in urban areas with a higher industrial release of Mn [210].
Gly acts as a Mn chelator, thus interfering with its metabolism. Severe Mn depletion can be another limiting step for the shikimate pathway in plants and bacteria, as Mn is a catalyst for EPSP synthase [211]. Disruption of Mn homeostasis can selectively affect Lactobacillus, which utilizes Mn as a protective mechanism against oxidative damage; thus, its requirements for Mn are much higher compared to other species [212], which can lead to its reduced number in the gut. This may be related to another neurotransmitter-linked microbiome-gut-brain mechanism. Several members of the Lactobacillus family can produce GABA, the main CNS inhibitory neurotransmitter implicated in several conditions such as anxiety or depression, possibly relevant also in PD, and alterations in GABA neurotransmission in PD have gained recognition in many disease-related symptoms [213]. Probiotic treatment with L. rhamnosus restoring central GABAergic function could be of interest as a potential novel therapeutic approach to (PD-related) depression [214].
It might seem contradictory that Gly exposure could be linked to PD pathogenesis involving Mn, given that glyphosate acts as Mn chelator. However, Gly impairs Mn metabolism by several mechanisms, and some of them could be related to dysbiosis. In the review of Samsel and Seneff it was proposed that, paradoxically, both Mn deficiency and Mn toxicity attributable to Gly can occur simultaneously [211] and that this disruption of Mn homeostasis leads to extreme sensitivity to its availability [215]. In liver, regulation of Mn levels in general vascular circulation is ensured by incorporation of its excess amount into BA, thus providing the gut bacteria repeated access to Mn, a process that is blocked by Gly due to its disruption of cytochrome P450 (CYP) enzymes, which are crucial for BA production [198, 216]. Mn accumulation in the liver can result in its transport via the vagus nerve to brainstem nuclei, causing neuronal damage leading to parkinsonism; this pathway could be related to Mn toxicity in the brainstem nuclei in the conditions of Mn abundance and the simultaneous presence of Gly [211].
Thus, on one hand, decreased serum levels of Mn, impairment of several Mn-dependent enzymes and its deficiency in brain areas apart from the brainstem are caused by Gly-mediated direct chelation of Mn as well as reduced bioavailability of Mn to the gut bacteria due to its failure to be incorporated into BA and its accumulation in the liver. On the other hand, the brainstem suffers from excess Mn levels. In the brain, Mn is preferentially localized in astrocytes; its overexposure can lead to Mn-induced excitotoxic neuronal injury by dysregulating astrocytic cycling of glutamine (Gln) and glutamate (Glu). In addition, reactive astrocytes are important mediators of Mn-induced neuronal damage by triggering neuroinflammatory responses [217]. Moreover, high concentrations of neuromelanin are found in both the SN and the locus coeruleus, and neuromelanin was also proposed to have a protective function via accumulating and retaining various amines and metallic cations, especially Mn [218]. As neuromelanin is related to dopamine (derived from Tyr) in the SN and to noradrenaline (derived from Trp) in the locus coeruleus, both dependent on shikimate pathway products, it was hypothesized that in case of Gly exposure and shikimate pathway inhibition in gut bacteria, neuromelanin is likely to be deficient, which would result in a reduced ability to temporarily store excess Mn in the brainstem nuclei until it can be disposed of [211].
There was a critical response stating that the relationship between Gly and Mn may be exaggerated in several aspects based on epidemiological studies rather than experimental evidence, indicating correlation, not necessarily causation. Thus, a stronger level of evidence is needed for the future final call for action (a potential global ban on Gly) [219].
Inflammation: Alterations in immune system function and promotion of inflammation can be another consequence of changes in gut microbiota. The effects of Gly and GBHs on immune function in different animals and isolated immune cells were recently reviewed, and it was concluded that many of them are mediated via changes in gut microbiota, with multiple proinflammatory effects (especially in fish) [220].
Other pesticides and toxins –MPTP, chlorpyrifos, permethrin
Several studies have also shown gut microbiota alterations in the MPTP-induced PD mouse model, such as decreased abundance and diversity with an increase of Ruminococcus, Parabacteroides, and Parasutterella genera and a decrease of Coriobacteriaceae, Flavonifractor, Lachnospiraceae, Lactobacillaceae, and Rikenellaceae (overall dysbiosis was associated with disturbance of the DA, kynurenine, and 5-HT metabolic pathways) [221], a change in the abundance of Lachnospiraceae, Clostridiales, and Proteobacteria (decreased) and Erysipelotrichaceae, Prevotellaceae, and Erysipelotrichales (increased) [222], a decrease in the Bacteroidetes phylum, the Lactobacillales order, and the Prevotellaceae family [223] or a decrease in the phylum Firmicutes (order Clostridiales) together with an increase in the phylum Proteobacteria (order Turicibacterales and Enterobacteriales) [224], with many findings showing consistency with observations in PD humans (increased abundance of Proteobacteria [179] and Enterobacteriales [225]).
An alteration of the gut microbiota composition (a decreased abundance of Firmicutes and Bacteroidetes) was also described in animals exposed to other pesticides, such as chlorpyrifos [226] and permethrin [227]. The first study was also validated in an in vitro SHIME model mimicking the human intestinal environment and demonstrating an increase in the numbers of Enterococcus and Bacteroides as well as a decrease in the numbers of Bifidobacterium and Lactobacillus after chronic exposure to low levels of chlorpyrifos [228].
Additional epidemiological data from a Norwegian cohort study revealed a potential influence on infant gut microbial function during a critical developmental window by uptake of low level pesticides from breast milk of a mother exposed to toxicants [229]. In detail, several Lactobacillus species were lower in abundance in samples from infants with relatively “high” vs. “low” toxicant exposure. As already discussed, Lactobacillus in general appears to be a genus that is highly prone to the effects of pesticides and environmental toxins.
Taken together, pesticide-induced dysbiosis and the shift to putative pro-inflammatory gut microbiota composition in general can (further) disrupt epithelial integrity, leading to gut leakiness, innate immune activation and possibly systemic inflammation [230–232]; moreover, there are some mechanisms unique to particular pesticides that lead to alterations of certain metabolic pathways or interference with neurotransmission.
Oral microbiome and the link to pesticides exposure
The oral microbiome has been studied far less compared to its gut counterpart, yet it might represent another overlooked missing piece in the overall picture of how environmental exposures impact human microbial communities. In a study of farmworkers occupationally exposed to organophosphates (insecticide azinphos-methyl), large-scale significant alterations of the oral buccal microbiota composition were detected, with extinctions of whole taxa suggested in some individuals (especially the Streptococcus genus) and long-lasting effects in repeated sampling (spring/summer –winter) [233]. Whether these changes could also be relevant in PD and might represent one of the early changes in microbiota composition occurring along the GI tract in response to ingested pollutants is not clear yet and requires further study.
The nasal microbiome, for instance, was shown to be relatively uninformative as a source of biomarkers for PD/iRBD (idiopathic REM Sleep Behavior Disorder, most relevant prodromal marker of PD) [234].
Microbiota mediate the effects of pesticides
Pesticide-induced dysbiosis represents only half of the reality; on the other side of the coin are multiple ways that the gut microbiota enable and modify the effects of pesticides by influencing their availability and metabolism, including many systemic pathways mediated by microbial metabolites and products.
Microbiota: presence required for pesticide toxicity, composition mediates toxin effects
It is widely agreed that consistent changes appear in the gut microbiota composition related to PD. However, this poses a chicken or egg question—does the underlying α-syn pathology affecting the ENS cause PD-related constipation, which in turn leads to dysbiosis, or do the alterations in gut microbiome due to multiple environmental factors lead to inflammation, α-syn accumulation, changes in the ENS, subsequent constipation and ascending PD pathology? Some hints come from animal models. In an α-syn overexpressing mouse model, germ-free or antibiotic (ATB)-treated animals were protected against neuroinflammation and motor dysfunction, whereas transplantation of stool from PD patients to these germ-free α-syn transgenic mice enhanced PD-like motor deficits and CNS pathologies significantly compared to microbiota transplants from healthy human donors. This suggests that gut bacteria are involved in PD pathogenesis to a high degree, and that dysbiosis alone can trigger PD pathology in a genetically susceptible host [163].
In the past, caution was raised due to the surprisingly high success rate of microbiome transfer experiments in germ-free animals in other disease models besides PD [235]. Whether the role of the gut microbiome was not being overstated and causality inferred in a false positive way was questioned. However, more studies followed in line with the first findings, and they indicated that microbiota act as an interface between the host and environmental exposures, and their presence and composition can alter the effects of pesticides both for better and for worse. Similar to the findings of Sampson et al. proving that fecal transplants from human PD subjects enhanced PD pathology in α-syn overexpressing transgenic mice (without any other external toxin exposure) [163], Sun et al. demonstrated that fecal transplants from MPTP-induced PD mice caused impaired motor function and decreased striatal dopamine and serotonin levels in control mice. Moreover, FMT from control mice to MPTP-induced PD mice improved their gut microbial dysbiosis and exerted neuroprotective effects, as evidenced in reduced motor impairment, elevated DA levels, reduced loss of DA neurons and restoration of 5-HT synthesis. The mechanisms underlying these beneficial effects were decreased activation of brain microglia and astrocytes and lowered fecal concentrations of SCFA, all mediated by inhibition of the TLR4/TBK1/NF-κB/TNF-α signaling pathway, thus suppressing neuroinflammation and gut inflammation [224]. Another study in a PD mouse model showed that ATB-induced microbiome depletion was protective against MPTP-induced DA neurotoxicity in the brain via the gut-brain axis [236]. Rotenone models yielded results consistent with previous studies—chronic rotenone treatment changed the gut microbiota composition significantly and caused an increase in intestinal permeability in conventionally raised mice, whereas no disruption of intestinal permeability was seen in germ-free mice, which again highlights the role of gut microbiota in regulating barrier dysfunction and motor deficits in PD [171].
These findings support the role of the microbiome in PD pathogenesis not only in transgenic animals with α-syn overexpression, but also in the context of exposure to environmental toxins. The gut microbiota is not only required for its neurotoxicity but also for mediating the effects of pesticides on intestinal permeability and the development of motor symptoms [171].
Mechanisms involved in microbiome-pesticide-host interactions
Huang postulated a hypothesis of “gut microenvironment baseline drift”, stating that the gut microbiota is a temporarily combined cluster of species sharing the same environmental stresses for a short period, which would change quickly under the influence of different environmental factors [237]. On the one hand, gut physiological properties and microbiome composition may be altered by hazardous environmental factors, such as pesticides [29], as shown in the previous section (4.2). On the other hand, the microbiome can adapt to environmental insults for improved survival by altering its composition, increasing the number of mutations in its genome or generating metabolites and products inducing various cascades that can be either beneficial (SCFA) or detrimental for the host (proinflammatory cytokines including IL-1b) [237].
Leaky gut: enabling/increasing pesticide toxicity: Besides the outcomes of dysbiosis, such as leaky gut, intestinal inflammation and α-syn misfolding, contributing to PD on their own [125, 145], there is one more important consequence to mention: intestinal hyperpermeability is likely to expose the intestinal neural plexi to toxins, including pesticides [238]. Two major barriers should prevent neurotoxins from entering the CNS: the BBB and the intestinal wall in the absence of gut dysbiosis. Once dysbiosis is present, it increases the permeability of the intestine (leading to increased entry of neurotoxins) and the BBB as well, enabling deleterious effects of neurotoxins, including pesticides, on the CNS, leading to neurodegeneration [239]. Pesticides therefore create a mechanistic self-perpetuating vicious cycle in which they first cause gut dysbiosis leading to inflammation and impaired intestinal barrier, which in turn allows increased exposure to pesticides (and other environmental pollutants and putative pathogens as well), further enabling and increasing their toxicity.
Xenobiotic metabolism: Gut microbiota play a crucial role in the degradation and metabolism of xenobiotics, i.e. chemicals to which an organism is exposed that are not naturally present or produced in that organism [240]. The gut microbiome can change the pharmacokinetics of xenobiotics (including pesticides) in multiple steps, affecting their bioactivity and toxicity, e.g., by inducing direct chemical modification leading to increased metabolism or bioactivation and reversing the modifications imparted by host detoxification pathways or by regulating host gene expression [241, 242]. In PD, increased metabolic activity of the microbial pathways involved in the degradation of xenobiotics, including herbicides (namely atrazine), was found in stool samples compared to controls [175]. Several Proteobacteria in the human gut microbiome have been found to degrade glyphosate using the carbon–phosphorus lyase pathway [197].
Metabolic/endocrine pathways: It was discovered that pesticides can induce obesity and endocrine disruption, all mediated by the gut microbiome and its metabolites [243]. There are many converging pathways involved, including insulin resistance, obesity, lipid metabolism, atherosclerosis, appetite changes and others [28, 237]. Although studies on the mechanisms by which pesticides can affect human metabolism through influencing the gut microbiota were not directly focusing on PD, given that obesity, metabolic syndrome and type 2 diabetes are risk factors for PD [184] and insulin resistance is one of the hallmarks of PD [244], these factors may emerge as important indirect effects of pesticide exposure and hidden drivers of PD pathogenesis beyond direct DA neurons damage.
Regarding specific toxin PD-models, decreased Prevotellaceae and increased Lactobacillaceae and Bifidobacteriaceae were found in a rotenone-induced rat model of PD [173], and these changes have been associated with reduced serum ghrelin [177, 180], a gut hormone regulating nigrostriatal dopamine function with the proposed ability to attenuate neurodegeneration in PD [181]. In this aspect, PD patients have been reported as having impaired ghrelin secretion, which may increase the vulnerability of the nigrostriatal neurons and contribute to the development of GI symptoms in PD patients, as ghrelin promotes gastrointestinal motility [245].
An overview of pesticides and the microbiome-gut-brain axis interactions and contributions to the pathogenesis of PD is displayed in Fig. 1.
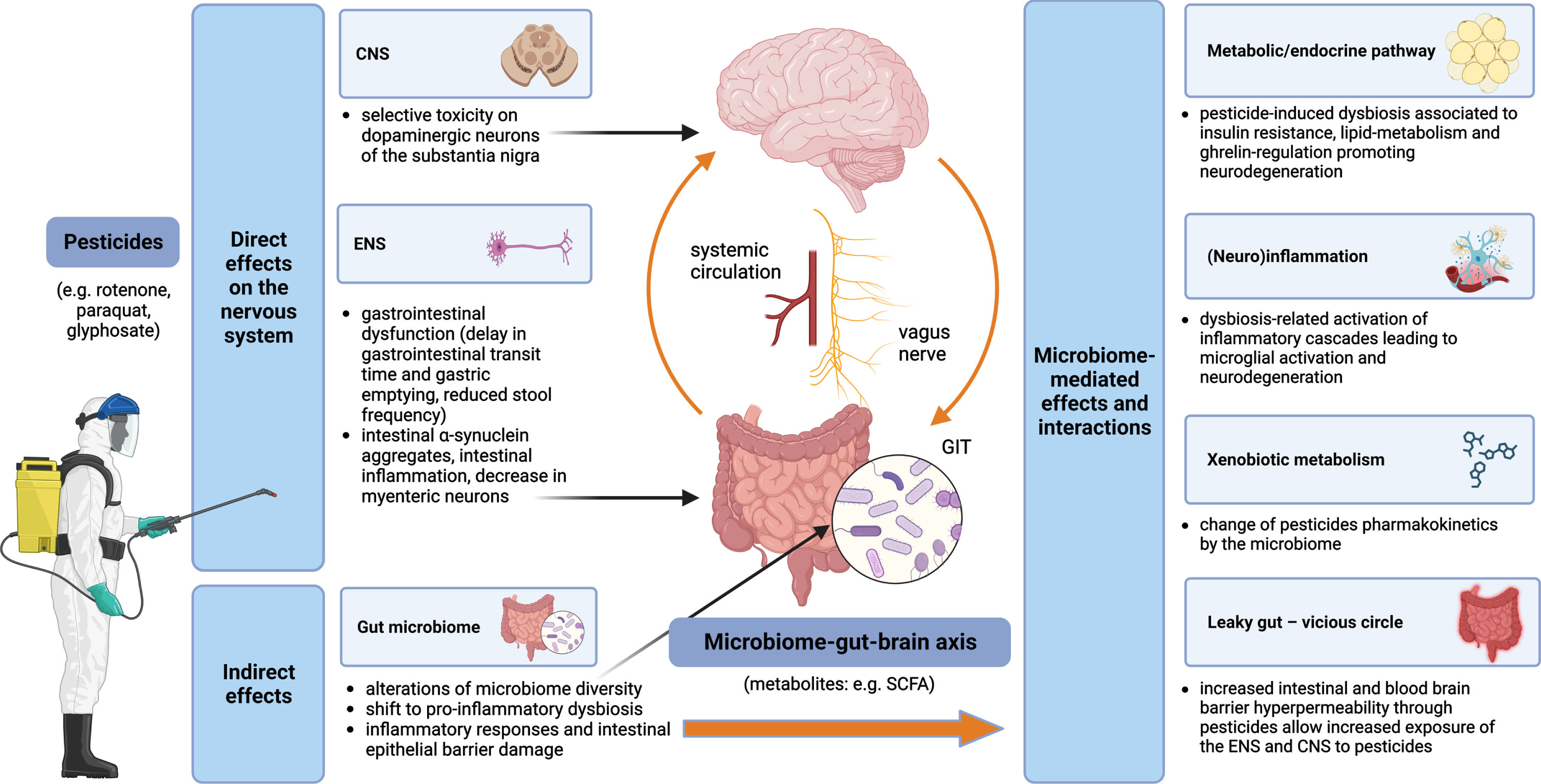
Pesticides and the microbiome-gut-brain-axis in PD pathogenesis –convergent pathways. Pesticides exert both direct and indirect effects on the nervous system. Direct mechanisms include toxicity on dopaminergic neurons of the substantia nigra in the CNS and impairment of the ENS, while indirect mechanisms are related to alterations of the gut microbiome with multiple subsequent effects, such as leaky gut (leading to self-perpetuating increased exposure of the ENS and CNS to the pesticides), (neuro)inflammation and metabolic/endocrine disruption (mediated, e.g., by bacterial metabolites); gut microbiota can modify the effects of pesticides by changing their pharmacokinetics. CNS, central nervous system; ENS, enteric nervous system; GIT, gastrointestinal tract; SCFA, short-chain fatty acids.
GAPS AND FUTURE DIRECTIONS RELATED TO PESTICIDES AND THE MICROBIOME-GUT-BRAIN AXIS
Brain-first vs. body-first PD
Two basic PD phenotypes have been proposed in regards to the trajectory of disease progression, as introduced in the section on prodromal PD in general, the so-called brain-first PD and body-first PD [132], with the opposite origin site of α-syn misfolding and subsequent spreading (brain-first: descendent trajectory starting in the CNS, body-first: ascendent trajectory starting in the gut—the ENS/PNS). To acknowledge possible oversimplification, this model might not explain the full clinical phenotypic variability of PD (which is currently considered more a spectrum of diseases than just one unit [246]). Moreover, it might not be relevant for genetic forms of PD with almost no or only limited α-syn pathology (including, for example, the PRKN or LRRK2 mutation) and might be of questionable relevance to genetic forms of PD in general (as many have atypical clinical phenotypes, although some might fit into this model; LRRK2-PD patients mostly exhibit a brain-first PD profile, while patients with GBA variants typically resemble a body-first profile, and SNCA-variant carriers are equally distributed between both subtypes [247]). However, it may still be useful in cases of idiopathic PD (accounting for almost 90% of PD worldwide), presenting an interesting research framework to study the disease progression.
It was shown that the body-first group has a greater colon transit time and greater colon volume compared to the brain-first group, indicating more constipation [248]. Thus, a substantial contribution of the microbiome-gut-brain axis to PD pathogenesis from the very beginning seems to be plausible in the body-first subtype. Evidence for specific involvement of either pesticides or the microbiome in the brain-/body-first PD subtypes is currently limited, although some observations have already been made. Interestingly, it was shown that pesticide exposure is more common in PD cases with RBD and symmetric parkinsonism, representing the body-first subtype. This suggests that ingested pesticides may have a stronger propensity to trigger enteric pathology rather than olfactory bulb pathology [249].
As for microbiome changes in PD, the broad heterogeneity of findings has a number of possible reasons [250]; however, brain-/body-first phenotypes were not taken into account and could perhaps explain part of the inconsistencies [251]. It could be expected that PD-related changes in microbiota composition would be more pronounced in the body-first group. Although we do not have such direct evidence yet, as previous studies focused on PD mostly as a single group without differentiating between the proposed subtypes, the first indications come from studies of iRBD patients (as a proxy for prodromal body-first PD). More than 75% of the identified bacteria differing between PD and controls were qualitatively similar to the differences between iRBD and controls [234, 252].
Overall, it seems that both pesticides and microbiome-related contribution to PD pathogenesis is more linked to the body-first phenotype, although these conclusions are still preliminary, and further studies are definitely needed to gain deeper insight into the differences between these two PD subtypes.
Gene-environment interactions
An example of potential gene-environment interactions is the crosstalk between LRRK2 protein and the pesticides rotenone [253–255] and paraquat [256, 257], as well as other environmental toxicants, such as solvent trichloroethylene [258] and manganese [259, 260]. Pathological elevation in LRRK2 kinase activity induced by environmental toxicants resembles LRRK2 gain-of-function mutations; therefore, pharmaceutical inhibition of LRRK2 by small molecule kinase inhibitors [261] could be appropriate after certain exposures [42, 262]. Taken together, it can be hypothesized that LRRK2-mutation carriers might be particularly vulnerable to the above-mentioned pesticides. Another example of gene-environment interaction that promotes risk for PD via alterations in immune responses can be a single nucleotide polymorphism in the major histocompatibility complex class II (MHC-II), which increases susceptibility to PD through the presentation of pathogenic, immunodominant antigens and/or a shift toward a more pro-inflammatory CD4 + T cell response in response to specific environmental exposures, such as pyrethroid exposure [263]. Defective cellular protection against oxidative stress represents another mechanism. PD risk from paraquat exposure was high in individuals with homozygous deletions of the genes encoding glutathione S-transferase [264]. Scavenger capacity also plays a role: butyrylcholinesterase (BChE, a known bioscavenger for pesticides) decreases PD risk, while K-variant BCHE reduces the serum activity of BChE and results in increased PD risk in pesticide-exposed individuals, particularly in case of insecticides, such as organophosphates and carbamates [265].
Genetic background can also influence pesticide-microbiome-host interactions, although findings are still rather scarce. Mutations of the HFE protein (High Fe2 +, a critical human regulator of cellular iron uptake) are associated with increased iron accumulation, including the brain, and the commonly occurring H63D variant is considered as a disease modifier in several neurodegenerative diseases [266]. HFE mutant mice showed reduced expression of tyrosine hydroxylase in the SN; however, they presented with alterations in the gut microbiome profile contrary to PD pattern and were resistant to paraquat toxicity, altogether providing additional support for HFE genotype being a disease modifier for PD as well as a model to study mechanisms of resistance to neurotoxicants [267].
Many gene-environment interactions were studied in isolation. In future research, it will be important to develop comprehensive and systematic approaches to these complex relationships [42], such as studying genetic risk factors modifying pesticide-microbiome interactions in PD.
TARGETING PESTICIDES AND THE MICROBIOME-GUT-BRAIN AXIS: POSSIBILITIES FOR TRANSLATION
Given the current unavailability of causal disease-modifying treatment for PD while facing a Parkinson pandemic, especially in areas with rapid industrialization, it is important to view pesticides, the microbiome and their crosstalk as potential targets for preventive strategies. As the global prevalence of PD is projected to double by 2040 [268], all efforts should be made to decrease the burden through the lens of modifiable factors [5]. Where feasible, PD should be prevented by reducing and in some cases eliminating the use of chemicals known to increase the risk of PD, pesticides in particular [7]. On the other hand, any final call for action has to stand on a solid scientific foundation concerning the causative role of pesticides in PD pathogenesis, keeping in mind the consequences of the potential banning of glyphosate or other pesticides in a global perspective [219]. Fighting neurodegeneration must go hand in hand with fighting starvation in developing countries, so that solving the first problem (by the potential banning of multiple pesticides) does not deepen the second (by subsequent lower food production), especially given that pesticides are just one piece of the mosaic in PD pathogenesis and even banning all of them globally would not lead to the eradication of PD.
Although many mechanisms of pesticide-induced neurodegeneration have already been discovered, a lot is still unknown, posing a challenge for bulletproof large-scale recommendations concerning pesticide use or banning. In a “manifesto” titled as an environmental agenda to prevent PD, De Miranda et al. compiled research priorities from both basic and translational perspectives—to model complex environmental exposure and identify its relevant concentrations and routes of entry, to examine gender differences, gene-environment interactions and modifying effects of microbiome and diet, to track global PD incidence and its change with a special focus on populations with high exposure burden and to develop reliable biological markers of exposure and use them as early as in the prodromal phase [42].
The outright banning of pesticide use is currently not feasible due to food security concerns and the lack of viable alternatives for large-scale replacement [269]. One possible way out and a promising alternative to reduce or replace the use of agrochemicals is the use of biopesticides and genetically modified organisms, microorganisms that target specific pathogens, biochemical pesticides that control insect behavior and plant-incorporated protectants [270–272]. However, there are several challenges for a large-scale utilization of biopesticides to become a reality: their cost-effectiveness, farmers’ awareness, eventual incompatibilities between pesticides and microbial inoculants and safety to non-target organisms (including humans) [269]. Last, but not least, it is important to promote international cooperation to support the monitoring capabilities of developing countries, which usually lack the expertise and resources to implement effective monitoring programs for pesticide contaminants in both drinking water and food [273].
Still, even in the case of potential pesticides banning or an instant global transition to biopesticides, the long latency between these restrictions and any evident protective effect on the prevalence of PD has to be expected. Yuan et al. found high pesticide exposure among US farmers to be related to prodromal features of PD, such as dream-enacting behavior (indicating iRBD) with a time gap up to 40 years, which means the interval between pesticide exposure and the onset of manifest PD can be even up to 5 decades [49, 274], and early adulthood might be an etiological window of particular interest [275]. The results of current changes might take decades to be harvested, yet this fact should not serve as an excuse for resignation, but instead, as a motivation to search for alternative preventive measures that can be applied in the meantime.
Except for global recommendations to decrease the use of pesticides with a known connection to PD, there are some smaller but not unimportant steps to take for those who cannot avoid pesticide exposure. First, the use of protective tools and hygiene practices modified the association between pesticides and PD in epidemiological studies. Neither pesticide was associated with PD among protective glove users [276, 277], and a longer delay between the event of high pesticide exposure and subsequent washing with soap and water further increased the risk of dream-enacting behaviors [49]. Thus, taking personal protection equipment and hygiene seriously should be emphasized in agricultural workers to prevent occupational exposure, and using some forms of personal protection could also be advised to people who live in areas with high pesticide use to lower the environmental burden. Second, new innovative approaches (such as the utilization of N-acetyl cysteine, vitamin C, vitamin E, cyclophosphamide, or probiotics) are proposed for the treatment of acute herbicide poisoning where no proven antidote exists (paraquat, glyphosate) [278]. If the benefit is confirmed and the safety profile is acceptable, it would also be worth considering potential benefits of these approaches in terms of chronic exposure.
The other side of the coin is placing the focus on the microbiome-gut-brain axis and designing future preventive and therapeutic strategies to increase its resistance against pesticides. Concerning dietary habits and PD, higher intake of N-3 polyunsaturated fatty acids (PUFAs) was inversely associated with PD risk (possibly related to the ability of N-3 PUFAs to limit the inflammatory response). Moreover, fat intake modified the associations of PD with the pesticides paraquat and rotenone, suggesting that a diet low in PUFAs or high in saturated fat may increase vulnerability to pesticides and increase PD risk in case of exposure [279]. Developing specific approaches to strengthen the microbiome in PD through exercise, diet modification, prebiotics, probiotics, postbiotics, antimicrobial agents or fecal microbiota transplants is in the scope of several recently published reviews [28, 281]. These strategies could be of relevance, especially in professions and areas where pesticide exposure cannot be avoided, but due to a generally safe profile they might be beneficial also on a large-scale population basis (at least the lifestyle modifications). A summary of potential preventive measures related to both pesticides and microbiome is outlined in Fig. 2.
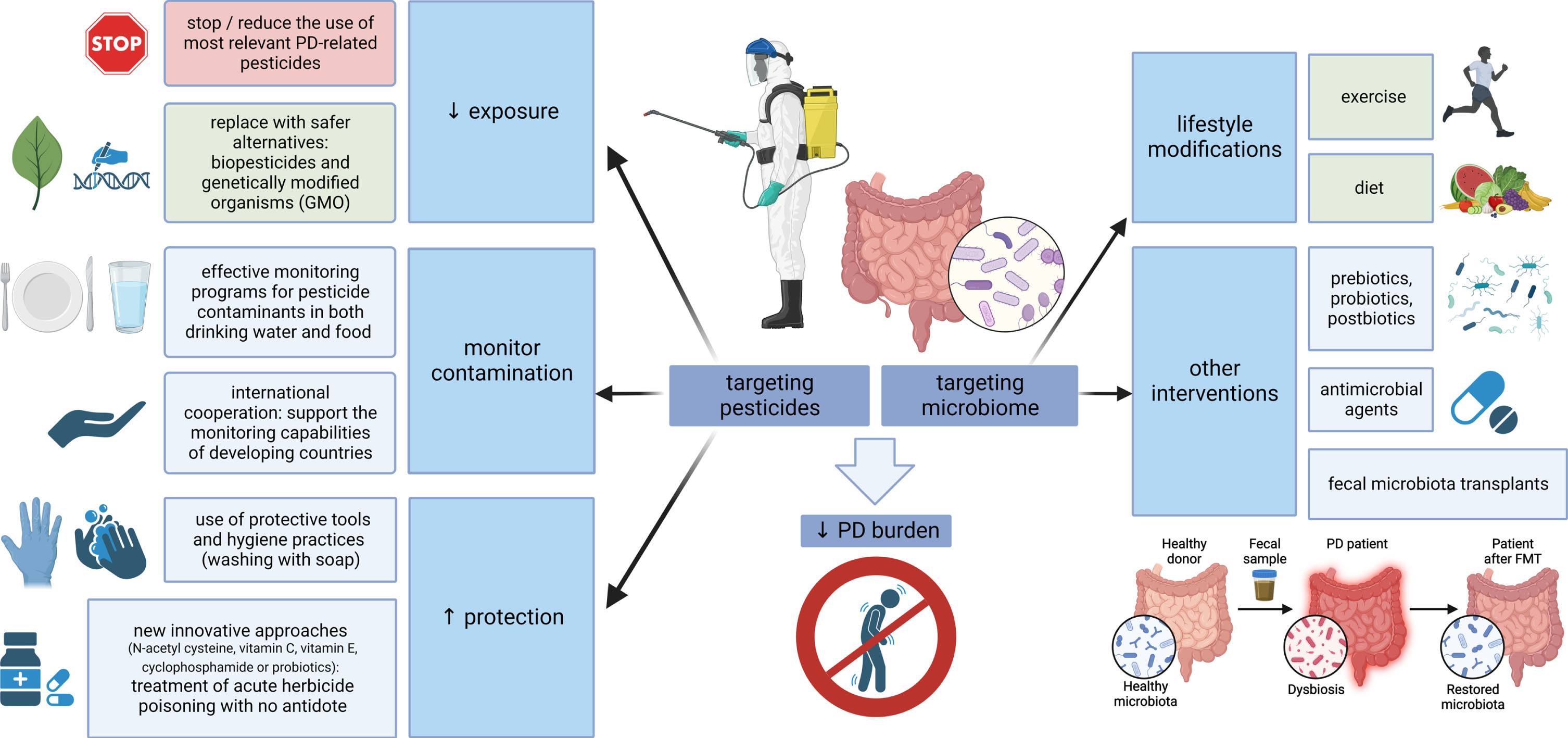
Targeting pesticides and the microbiome-gut-brain axis. Multiple measures are at hand to reduce the global burden of PD, aimed at both pesticides and microbiome. Where possible, the use of PD-relevant pesticides should be reduced and replaced by safer alternatives; effective monitoring should control food and water contamination, and protective measures, such as the use of personal protective equipment or new approaches for the treatment of poisoning, should be emphasized where pesticide exposure is inevitable. On the other hand, strategies aimed at strengthening the microbiome could be used on a large scale (general lifestyle modifications, such as exercise and a healthy diet), while more targeted interventions, including the use of pre/pro/postbiotics, antimicrobial agents or fecal microbiota transplants, could be relevant in specific conditions associated with pesticide exposure. PD, Parkinson’s disease; GMO, genetically modified organisms.
CONCLUSIONS
The increasing global burden of PD draws our attention to potentially modifiable causes that would open the door for preventive measures. Historical evidence of the existence of PD centuries before synthetic pesticides and industrial solvents were manufactured and extensively used argues against their having a sole causative role. However, current epidemiological studies linking chronic occupational or environmental pesticide exposure with higher PD prevalence [282], records of PD development after acute high-dose pesticide intoxication, as well as animal PD models where the disease initiation and progression follows experimental pesticide exposure alone provide strong proof that pesticides may be significant contributors in at least a subset of PD cases.
Although many risk factors and disease modifiers were assessed independently in the past, it is clear today that influences of the environment and lifestyle, the gene-environment and environment-environment interactions are enormously complex and deserve focused interest to untangle hidden convergent pathways and mutual influence on PD pathogenesis. Air pollution emerges as one of newly recognized factors increasing PD risk and acting through multiple similar mechanisms as pesticides: direct neuronal toxicity, induction of systemic inflammation leading to CNS inflammation and alterations in gut physiology and the microbiome [283]. Various environmental factors might act synergistically, as illustrated by the example of paraquat and traumatic brain injury: both increase the risk of PD independently, but combined exposure almost triples the risk of the disease [284, 285].
Pesticide-microbiome crosstalk is an important part of the overall picture. Many effects of both pesticides and microbiome converge in impairing intestinal barrier integrity, leading to gut and systemic inflammation, with microbial metabolites playing an important role in mediating multiple inflammatory and metabolic cascades, finally resulting in an accumulation of misfolded α-syn and neurodegeneration.
A lot is still unknown: the differences between body-/brain-first PD, the complexity of environmental pollution, the search for relevant and reliable biomarkers of toxicant exposure, understanding gender differences, genetic mechanisms of susceptibility and resistance, the multi-omics perspective of microbiome-mediated effects and many others. Importantly, even today we know enough to move one step forward to focus on preventive measures to soften the deleterious impact of pesticides and dysbiosis on the PD burden. Where possible, the use of dangerous pesticides with a clear link to PD, such as paraquat, should be discouraged and replaced by safer alternatives. In the meantime, the use of protective equipment should be emphasized, and new pharmacological or complementary dietary approaches should be considered in populations unable to avoid pesticide exposure. Last but not least: many microbiome-targeted strategies, such as exercise, diet, prebiotics, probiotics, postbiotics, ATB, small molecule drugs, phage therapy or FMT, are emerging. They represent promising tools with a safe profile not only for high-risk agricultural occupations, but also to induce positive changes and strengthen the microbiome in the general population. To conclude, although further research is needed to understand the pathophysiological background better, possible meaningful preventive interventions to fight the neurodegeneration and PD pandemic, such as reducing the use of harmful substances or promoting healthy lifestyle modifications, are at hand, which is hopeful particularly in the conditions of unavailability of causal disease-modifying treatment of PD that we still have to face.
Footnotes
ACKNOWLEDGMENTS
All figures were created with BioRender.com.
FUNDING
This work was supported by Slovak Research and Development Agency under contract no. APVV-22-0279 and by the Slovak Scientific Grant Agency under contract no. VEGA 1/0712/22 for the Slovak site.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.