
Abstract
Background:
A substantial body of research has examined the relationship between alcohol consumption and risk of Parkinson’s disease (PD).
Objective:
To provide an updated systematic review and meta-analysis of observational studies examining the relationship between alcohol consumption and risk of PD.
Methods:
Eligible studies comparing PD risk in ever vs. never alcohol drinkers were sourced from six databases. Outcomes were pooled using standard meta-analysis techniques. Separate female and male estimates were generated from studies reporting sex-specific data. Additionally, cohort studies stratifying participants by quantity of alcohol intake were integrated in a dose-response analysis.
Results:
52 studies were included, totaling 63,707 PD patients and 9,817,924 controls. Our meta-analysis supported a statistically significant overrepresentation of never drinkers among PD subjects; odds ratio (OR) for ever drinking alcohol 0.84 (95% confidence interval (CI) 0.76 – 0.92). A subgroup analysis revealed similar effect estimates in females and males. A further synthesis of seven cohort studies suggested a negative, dose-dependent association between alcohol and risk of PD.
Conclusion:
In the absence of a known neuroprotective pathway, there may be reason to doubt a true biological effect. The role of survivor bias, selection and recall bias, misclassification, and residual confounding requires consideration. Alternatively, observations might be attributable to reverse causation if those predestined for PD alter their alcohol habits during the preclinical phase. Major limitations of our study include high between-study heterogeneity (I2 = 93.2%) and lack of adjustment for key confounders, namely smoking status.
Keywords
INTRODUCTION
Parkinson’s disease (PD) is a common neurodegenerative disorder which probably stems from the interplay of genetic susceptibility and environmental exposures [1]. Epidemiological data has highlighted numerous putative risk factors [2]; however, the extent to which associations from observational studies represent true causal effects is contested. A substantial body of research has sought to determine the impact of alcohol drinking on PD risk. While the majority of observational studies have supported an apparent protective association, many smaller studies have lacked the statistical power and follow-up time required to draw firm conclusions.
To date, five meta-analyses [2–6] and one systematic review [7] are available on the subject. Though each supports a protective association—with the exception of one [5], whose findings for cohort studies were equivocal—the general consensus is that larger, prospective analyses are required. Since 2019, four more longitudinal studies (including two nationwide cohorts [8, 9]) have been published. Here, we aim to provide an updated systematic review and meta-analysis, with examination of potential sources of bias in the existing data.
METHODS
Database search
The terms ‘Alcohol’ AND ‘Parkinson disease’ were entered into the following databases: PubMed, OVID (Medline), Web of Science, Embase, Scopus, and preprint servers bioRxiv/medRxiv. Associated synonyms were added as both index and free text terms; see Supplementary Table 1 for a complete list. The final search was performed on April 19, 2022.
Inclusion criteria
We included both prospective and retrospective observational studies examining the relationship between premorbid alcohol consumption and PD risk.
Exclusion criteria
Papers published in a non-English language, abstracts, and duplicates were excluded. Where study populations overlapped, the larger of the two studies was selected [10–24]. Studies involving alternative causes of parkinsonism in patients or controls were excluded [25–28]. We required that control groups did not include blood relatives of patients [29, 30]. For a twin study by Wirdefeldt et al. 2005 [31], we only extracted data comparing subjects with unrelated controls. We specified that studies must explicitly report on premorbid alcohol exposure (“alcohol history” was accepted); those exploring current alcohol consumption were removed [32–35]. Papers focusing on alcohol dependency, reporting the age of first drink or mean quantity of alcohol intake were excluded as outcomes could not be standardized.
Study selection
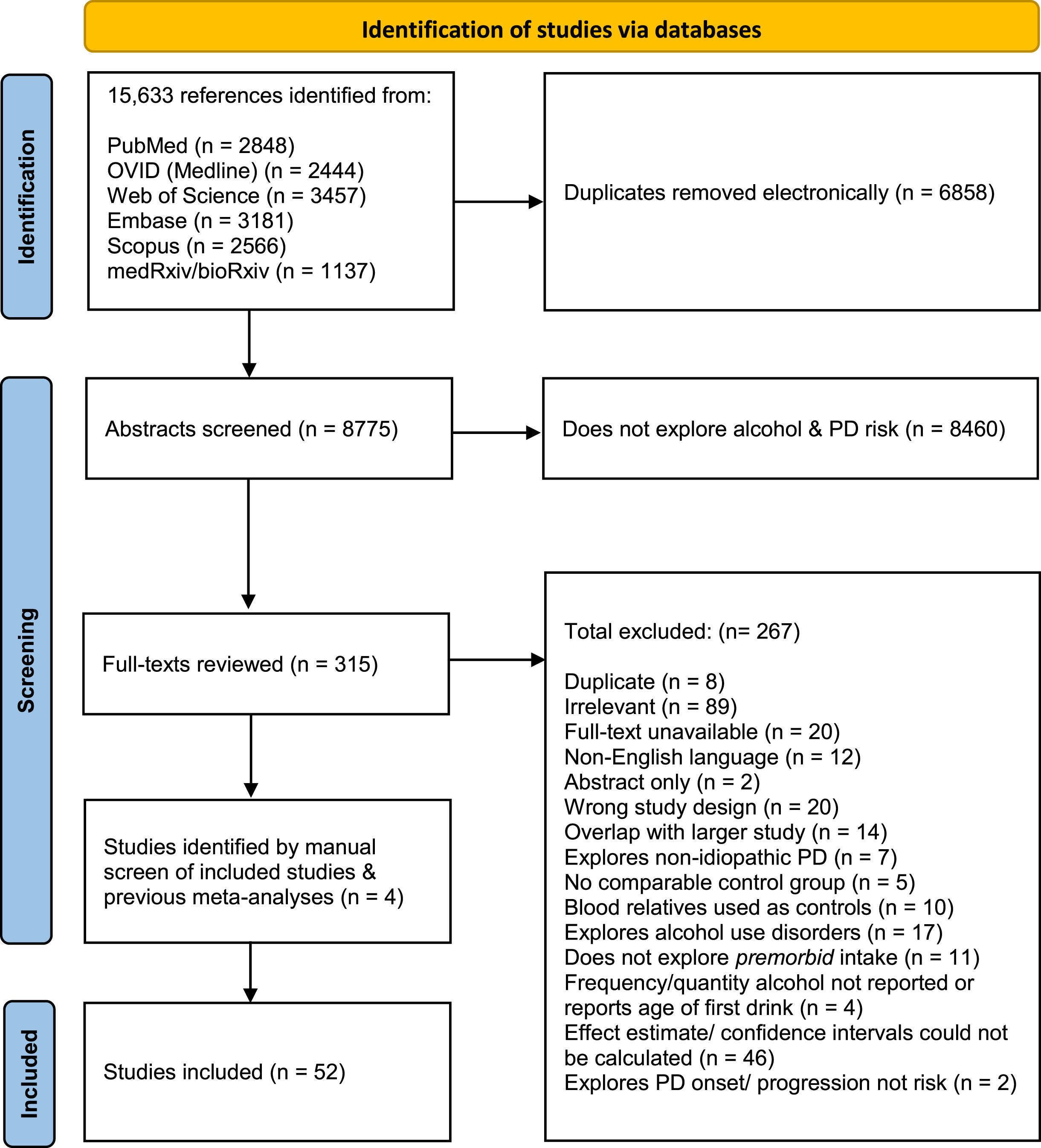
Our search returned 15,633 references, of which 6,858 duplicates were deleted. Following an abstract screen by researchers EM and HC, 315 full-texts were reviewed for eligibility. Where authors disagreed on the outcome, papers were re-reviewed and a decision was reached. The reference lists of included studies and previous meta-analyses were then manually screened. Four additional papers were identified this way. See Fig. 1 for a flowchart of the study selection process.
Data extraction
Information concerning study design, participant recruitment, exposure measurement, case ascertainment, and confounders were extracted by researcher EM. To integrate the multitude of methods employed to quantify alcohol consumption, we first grouped participants into binary drinking categories. ‘Never drinker’ referred to those in the lowest intake category reported in the paper, and the remaining participants were characterized as ‘ever drinkers’. Corresponding effect estimates and 95% confidence intervals (CI) with the highest available level of adjustment for confounding factors were extracted. Outcomes were reported as odds ratios (ORs) for case-control studies and as risk ratios (RRs), ORs, or hazard ratios (HRs) for cohort studies. As PD has a relatively low prevalence, these measures of association were treated as equivalent. Where effect estimates were not reported, we generated unadjusted ORs for case-control studies and RRs for cohort studies from the raw data. Beverage-specific calculations were beyond the scope of this paper. For the studies by Fall et al. (1999)[36] and Torti et al. (2020) [37], ORs were extrapolated from the number of wine drinkers as overall alcohol consumption could not be calculated. If available, outcomes stratified by sex were included. Finally, for cohort studies, outcomes according to specific quantities of alcohol intake were recorded. Study quality was assessed using the Newcastle-Ottawa Scale (NOS).
Statistical analysis
Individual study effect estimates were initially pooled by study design (case-control or cohort), then all together to produce an overall effect estimate, weighted by inverse variance. Separate female and male estimates were extracted from studies reporting sex-specific data. Heterogeneity was determined via the I2 statistic. As I2 was≥50%, a random effects model was employed for the main analysis. Publication bias was assessed by visual inspection of a funnel plot and application of Egger’s regression test.
Cohort studies stratifying participants by quantity of alcohol intake were integrated in a dose-response analysis. First, alcohol quantities were converted into grams/day, where a ‘drink’=13 grams of alcohol unless otherwise stated. The median dose for each drinking category was then obtained. Where the highest intake category was unlimited, we calculated an estimated median by adding the difference between the midpoint and upper limit of the previous category. We then generated a restricted cubic spline model with associated 95% confidence intervals. Knots were placed at 0, 10 and 38 grams/day.
We report our findings in accordance with PRISMA guidelines. Ethical approval was not required. All analyses were undertaken using Stata version 17.
RESULTS
Our search returned 38 case-control [11, 36–65] and 14 cohort [8, 66–77] studies, totaling 63,707 PD patients and 9,817,924 controls. See Supplementary Table 2 for full results of data extraction and quality assessment.
Ever vs. never alcohol drinking
Of the case-control studies, nine suggested a significant inverse association between overall alcohol consumption and PD risk. Most of the remaining studies (n = 28) showed no significant association, with only one paper reporting a positive association. For the cohort studies, half (n = 7) demonstrated a nonsignificant inverse association. Three reported positive associations, none of which were statistically significant. Notably, four of the more recent studies [8, 77]—including the two nationwide cohorts—demonstrated a significant protective association. However, this was based on our calculation of unadjusted risk ratios to allow binary comparison of ever vs. never drinkers. It should be noted that Kim et al. (2020) [8] and Peters et al. (2020) [77] both refute there being an association following a multivariate adjusted dose-specific analysis.
Overall, our meta-analysis supported a modest inverse association between overall alcohol consumption and PD risk. This was maintained when case-controls and cohorts were examined separately—OR 0.84 (95% CI 0.75–0.94) and OR 0.82 (95% CI 0.69–0.97) respectively—and in combination; OR 0.84 (95% CI 0.76–0.92). Sex-specific outcomes were considered in a subgroup analysis, revealing similar effect estimates for females and males; OR 0.81 (95% CI 0.61–1.08) and OR 0.76 (95% CI 0.58–1.01) respectively. See Figs. 2 and 3 for associated forest plots.
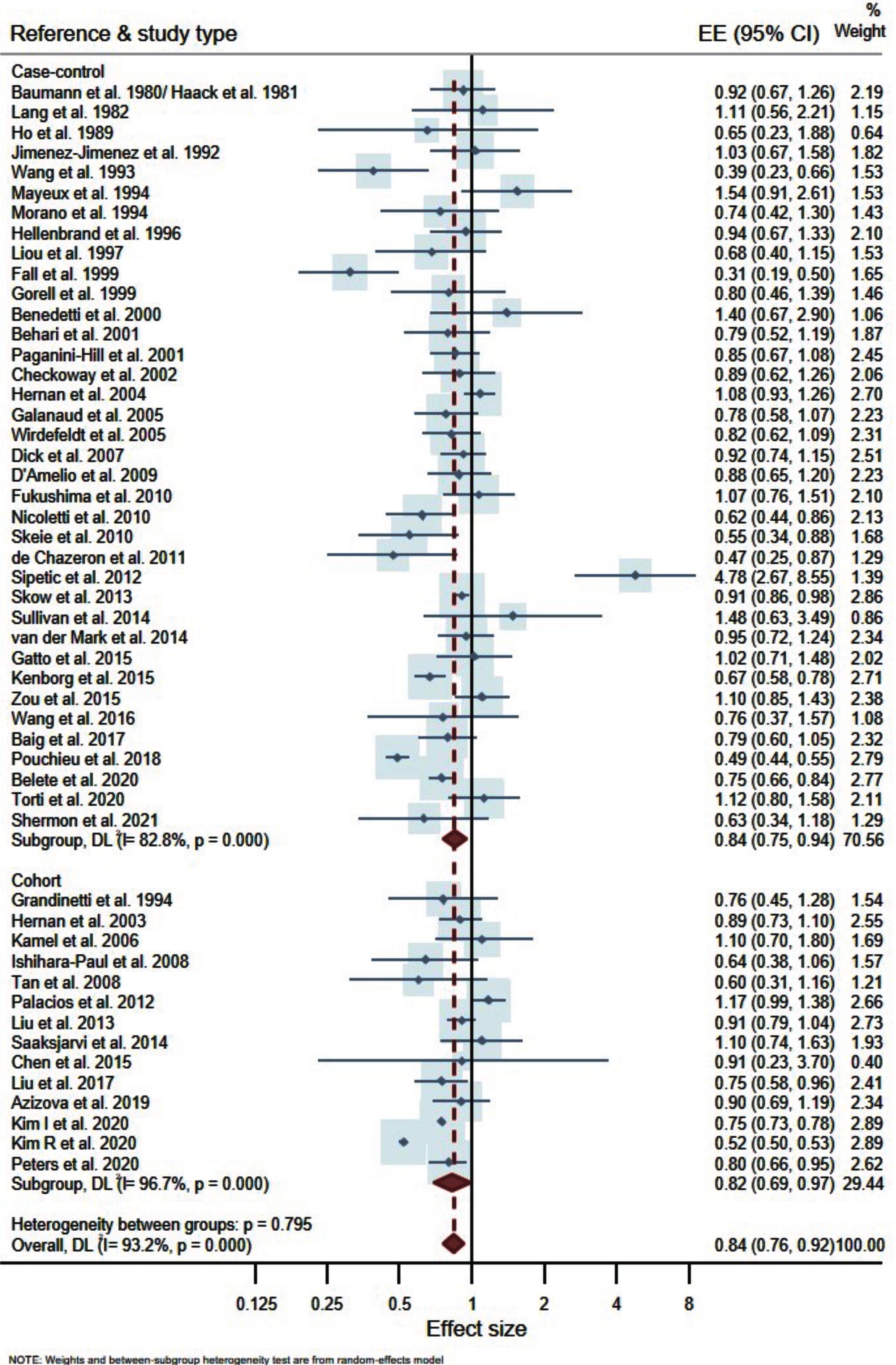
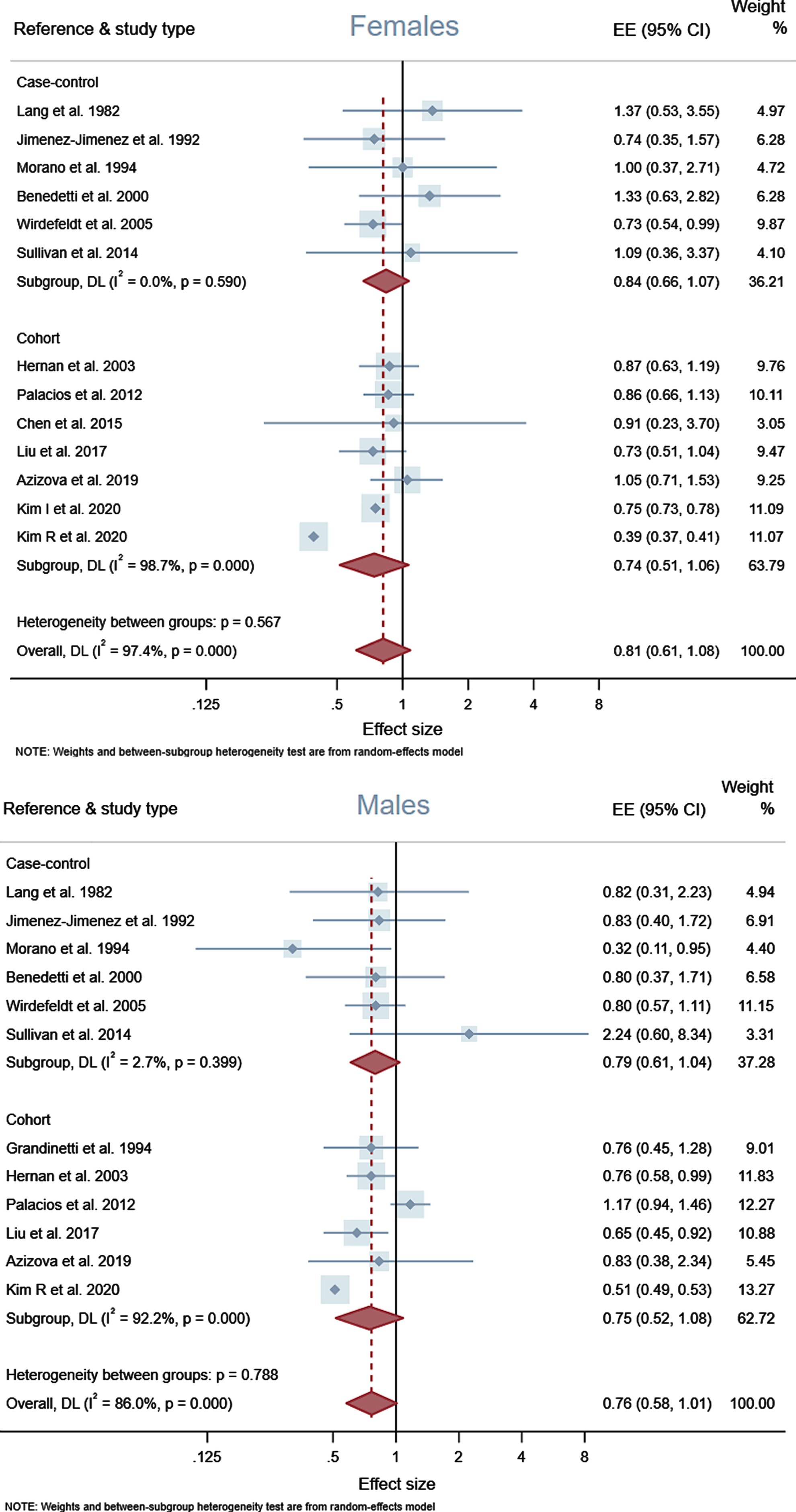
A lack of publication bias was suggested by a symmetrical funnel plot and application of Egger’s regression test (p = 0.261); see Supplementary Figure 1.
Dose-response analysis
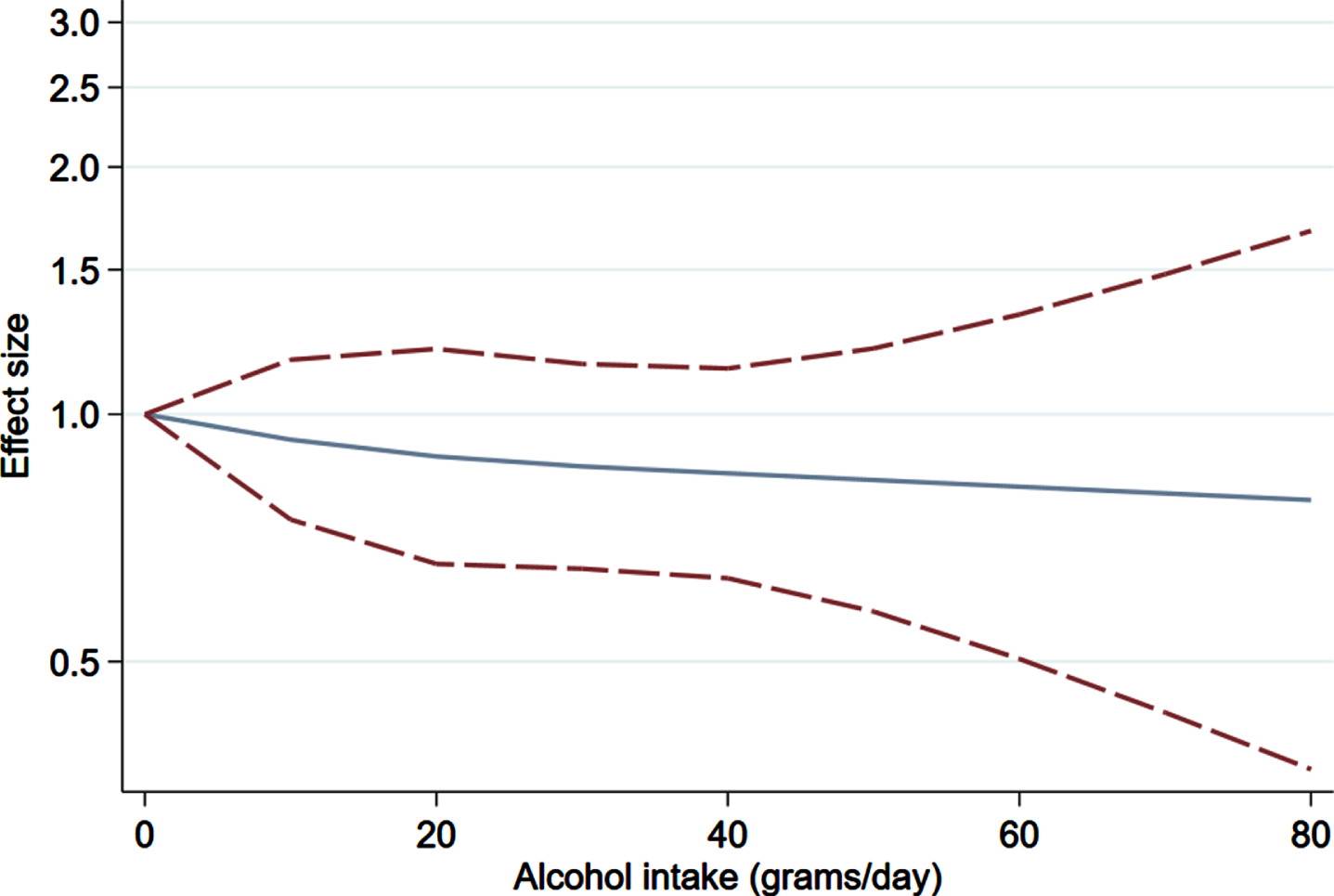
Peters et al. (2020) [77] was excluded from the analysis as they omitted required data for control participants. This left seven eligible cohort studies [8, 71–73] stratifying participants by quantity of alcohol intake, totaling 41,351 cases and 8,699,256 controls. For the paper by Kim et al. (2020) [8], we calculated unadjusted RRs as those published did not assume never drinker as the referent category. The corresponding restricted cubic spline model did not robustly support a significant dose-dependent decrease in PD risk with increasing alcohol consumption because the confidence intervals crossed the null at each dose level; see Fig. 4.
DISCUSSION
Our results show a modest but significant overrepresentation of never drinkers among PD patients, broadly in keeping with the consensus to date [2–6]. A subgroup analysis of males and females separately did not reveal differential association by sex, but the analyses were individually underpowered. A further synthesis of seven cohort studies suggested a trend for a negative, dose-dependent association of alcohol and PD risk. However, this relationship was not robustly supported as confidence intervals crossed the null at each dose level.
Bias
Although it is tempting to speculate that the observed inverse association reflects a potential protective effect, methodological explanations should be considered. Case-control studies in particular are susceptible to selection & recall bias, as well as misclassification of PD status and drinking history [7].
The behavioral link between alcohol and cigarette consumption poses a risk of residual confounding. In our main analysis, only 19% of effect estimates were adjusted for smoking habits. This is problematic given tobacco use is inversely associated with PD risk, in a manner shown to be dose-dependent and robust to multivariate adjustment [2, 79]. Although research this area is subject to similar biases as that of alcohol consumption, a causal link is possible given the reproducibility and magnitude of observed associations [13, 80–82]. The process by which tobacco use infers neuroprotection is currently unknown but several models are under investigation [81]. Quik et al. [83] propose that nicotine (and related compounds) might support nigrostriatal cell survival via stimulation of nicotinic acetylcholine receptors. Subsequent activation of calcium-mediated intracellular signaling pathways may ultimately promote immunomodulation and release of neurotrophic factors. Furthermore, nicotine has been shown experimentally to inhibit α-synuclein fibrillation [84] and trigger dopamine release [79]. Other cigarette constituents have demonstrated inhibition of the monoamine oxidase B (MAO-B) enzyme, resulting in reduced dopamine metabolism and modulation of otherwise neurotoxic cascades [83].
Additionally, the potential contribution of survivor bias warrants consideration. Survivor bias is introduced when an exposure (e.g., alcohol use) exerts differential health effects between cases and controls [85]. Plausibly, individuals who develop PD might have increased vulnerability to alcohol-related mortality than their non-PD counterparts. Crucially, alcohol in addition to PD must limit life expectancy to a greater extent than alcohol alone for survivor bias to operate. If so, PD patients (or preclinical cases) who drink alcohol would die earlier, exiting the population available for enrolment into studies [86]. This would lead to the erroneous conclusion that alcohol use is inherently less frequent in this population.
As a rare and late-onset disease, the study of PD lends itself to a case-control design, where subjects have, by definition, survived to the age of diagnosis. The majority of our observations are extracted from case-control studies, which are biased to preferentially select subjects with healthier premorbid lifestyles. Several studies attempted to mitigate this by enrolment of only newly diagnosed patients, and all but three specify exclusion of prevalent cases [36, 38–40]. However, a substantial risk of survivor bias will persist unless a prospective, longitudinal approach is adopted.
For cohort studies, establishing an appropriate baseline recruitment age is problematic. PD is rarely diagnosed under the age of 50, but has an indeterminate prodromal phase that could span decades, during which differential survivorship can occur [1]. Across the fourteen included cohort studies, only four [67, 76] had a mean recruitment age of < 50 years. Baseline measurements should ideally take place prior to first exposure; however, when quantifying alcohol consumption, this is rarely practicable. Complete follow up of all participants was best achieved in studies involving regular review of multiple information sources such as medical records, insurance databases, and death certificates [8, 75–77]. Those instead relying solely on infrequent telephone/mail questionnaires were likely to introduce survivor bias if deceased or disengaged participants ‘fall through the net’ [67, 74].
It is theoretically possible to determine if there exists an unrecorded excess of PD patients in those who succumb to alcohol-related disease. This would, however, require rigorous comparison of age-specific mortality causes/rates between drinkers and nondrinkers. Findings could then be cross-referenced with PD status. This is resource intensive; none of our included studies attempted such analysis. Furthermore, even with the above measures, one could never definitively determine the number of drinking individuals in a cohort who died before PD declared itself. Such a trend might be exaggerated if PD were to be under-reported in populations with higher drinking rates, where alcohol-related diseases ‘displace’ less acute neurological ailments on death certificates [38, 87]. Sourcing information on dead participants from medical records or relatives is prone to similar information biases [85]. Moreover, early PD may be missed entirely if alcohol drinkers are less inclined or resourced to seek a diagnosis. Thus, the true influence of survivor bias is difficult to definitively measure or control for.
Premorbid personality and reverse causation
An extensive literature already exists regarding the ‘premorbid personality’ hypothesis [35, 89]; that PD patients are predisposed against addictive behaviors due to a reduced internal reward response. As PD is characterized by depletion of dopaminergic circuits, it is posited that individuals might alter their lifestyle before motor symptoms become apparent [90, 91]. For this reason, we limited our analysis to studies that explicitly enquired about premorbid alcohol use. However, given PD’s relatively long prodromal phase [92], our findings might still be attributable wholly or in part to reverse causation.
Causality
While it would be inadvisable for any individual to increase their alcohol intake to reduce their risk of PD, a demonstrable neuroprotective link would be illuminating for etiological research and development of therapeutic targets. Several candidate alcoholic ingredients have been proposed to attenuate oxidative stress in the nigrostriatal system, often viewed as a final common endpoint in PD pathophysiology [93, 94].
Urate, a potent antioxidant and iron chelator, is raised in the serum following alcohol consumption [95]. An inverse association between serum urate concentration and PD risk has been reported in epidemiological data [96–101], complemented by postmortem studies, as well as cellular and animal models (reviewed elsewhere [93, 102]). Interestingly, this relationship could explain the lower incidence of PD in beer-drinkers compared to other beverage-types [15, 95], although this has not been consistently observed [8, 71]. Though plausible, research in this area is susceptible to bias introduced by confounding and survivorship. Moreover, trials of urate-elevating therapies in PD have failed to clinically translate [103], while the health risks of hyperuricemia are non-trivial. Other alcoholic components investigated for neuroprotective properties include niacin [103], flavonoids such as resveratrol [104–106], and ethanol itself [107]. Nonetheless, proposed models remain in the theoretical stages.
Mendelian randomization
The association between alcohol consumption and PD risk can be investigated at the genetic level. Mendelian randomization (MR) offers an approach to determining causality where a research question is ill-suited to be explored in a randomized control trial (RCT). The technique uses genetic variants, typically single nucleotide polymorphisms (SNPs) determined from genome-wide association studies, as proxies for environmental exposures. The random distribution of alleles within a population is analogous to the randomization process of an RCT [108]. For example, SNPs strongly associated with alcohol use (and no confounders) can be selected and tested for correlation with a PD phenotype. A second regression can be run in the opposite direction to ascertain if genetic markers for PD risk are linked to alcohol consumption. Observed associations can therefore be inferred as unidirectional causality rather than reverse causation or confounding [109, 110]. Some studies have indicated a protective effect [109–111], although findings have not been replicable [112–114]. The major limitation of MR lies in the potential for horizontal pleiotropy, where an instrumental variable affects a disease outcome via multiple pathways. Furthermore, as they adopt a case-control design there remains the potential for survivor bias, although this can be examined in simulation studies [108].
Limitations
For the purpose of data integration, it was necessary to group alcohol habits very broadly; we were unable to capture more subtle distinctions with regards to beverage preference or fluctuations in drinking habits. Although all included studies examined premorbid drinking in some capacity, most did not comprehensively assess average lifetime alcohol consumption. This is particularly relevant given many self-defined nondrinkers are in actuality ex-drinkers who neglect to report previous consumption. This will undoubtedly have introduced a degree of misclassification. Studies also varied significantly in approach to participant sourcing, exposure measurement, case ascertainment and adjustment. This may in part account for the very high heterogeneity (I2 = 93.2%) observed. As outcomes were not reported uniformly, for a number of papers [8, 77] we calculated unadjusted effect estimates from primary data. As such, adjustment for key cofounders (namely age and smoking status) was lost. Finally, Caucasian and Asian participants were consistently overrepresented. Results may not be generalizable to other populations.
Conclusions
Our meta-analysis supported the ostensible connection between low alcohol consumption and increased PD risk. However, there remains reason for skepticism, especially in the absence of a convincing biological explanation. Future prospective studies should attempt to minimize survivor bias in study design, including a rigorous examination of age-specific mortality causes/rates. Furthermore, MR offers a complementary approach to exploring causality which mitigates some traditional methodological pitfalls, while introducing others.
Footnotes
ACKNOWLEDGMENTS
We would like to thank the over nine million individuals whose participation was necessary to produce this meta-analysis.
The Preventive Neurology Unit is funded by the Barts Charity.
CONFLICT OF INTEREST
The authors have no conflicts of interest to report.
AJN reports grants from Parkinson’s UK, Barts Charity, Cure Parkinson’s, NIHR, Innovate UK, Virginia Keiley benefaction, Alchemab, Aligning Science Across Parkinson’s Global Parkinson’s Genetics Program (ASAP-GP2) and the Michael J Fox Foundation. Consultancy and personal fees from AstraZeneca, AbbVie, Profile, Roche, Biogen, UCB, Bial, Charco Neurotech, uMedeor, Alchemab, and Britannia, outside the submitted work.