
Abstract
Accumulating evidence suggests that microglia and peripheral immune cells may play determinant roles in the pathogenesis of Parkinson’s disease (PD). Consequently, there is a need to take advantage of immune-related models of PD to study the potential contribution of microglia and peripheral immune cells to the degeneration of the nigrostriatal system and help develop potential therapies for PD. In this review, we have summarised the main PD immune models. From a historical perspective, we highlight first the main features of intranigral injections of different pro-inflammogens, including lipopolysaccharide (LPS), thrombin, neuromelanin, etc. The use of adenoviral vectors to promote microglia-specific overexpression of different molecules in the ventral mesencephalon, including α-synuclein, IL-1β, and TNF, are also presented and briefly discussed. Finally, we summarise different models associated with peripheral inflammation whose contribution to the pathogenesis of neurodegenerative diseases is now an outstanding question. Illustrative examples included systemic LPS administration and dextran sulfate sodium-induced colitis in rodents.
Keywords
INTRODUCTION
Parkinson’s disease (PD) is characterized by a significant loss of dopaminergic neurons in the substantia nigra (SN) along with immunopositive intracellular neuronal inclusions for α-synuclein (α-syn) in the midbrain [1]. Different mechanisms have been suggested to play an essential role in the pathogenesis of PD, including impaired mitochondrial function, autophagy, loss of trophic support, protein homeostasis dysfunction, and neuroinflammation. Since the original observation by McGeer et al. in 1988 showing reactive microglia in the SN of human postmortem PD brain tissue [2], evidence supporting an important role of microglia and inflammation in driving neurodegenerative events is overwhelming. For instance, at the genetic level, it has been shown that PD risk alleles likely alter the functioning of microglia-specific enhancers in the loci LRRK2 and FCGR2A, specifically through disrupting a SPIB-binding motif in the latter [3]. Mutations in GBA1, the gene encoding the lysosomal enzyme glucocerebrosidase, are considered the most significant risk factors for PD, which is believed to create toxic species of α-syn aggregates through defective lysosomal function [4]. A recent mouse brain cell atlas supports that Gba1 is mainly expressed by microglia and not by neurons [5]. A role for T cells is also implied by identifying specific major histocompatibility complex (MHC) haplotypes and non-coding SNPs in MHC genes as risk factors for PD [6]. Different animal models have consistently shown that early microglia activation may precede the death of dopaminergic neurons, suggesting that brain immune cells may play a leading role in the pathogenesis of PD. Pattern recognition receptors (PRRs) sense the environment by recognizing pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns molecules (DAMPs) [7]. Illustrative examples of PRRs are toll-like receptors (TLRs), nod-like receptors (NLRs), and triggering receptors expressed on myeloid cells-2 (TREM2) [7], whose selective activation is thought to generate either a proinflammatory or a disease-associated microglia (DAM) phenotype [8]. With the advent of single-cell RNAsequencing (scRNA-seq) of microglia under disease conditions, it has become evident that microglia may acquire an array of activation phenotypes much larger than originally believed including potentially protective microglia phenotypes (DAM) [8], deleterious microglial neurodegenerative phenotype (MGnD) [9] or yet to be defined (such as white matter-associated microglia (WAM) [10]). scRNA-seq from the murine midbrain identified a microglia subtype exhibiting typical pro-inflammatory features including enrichment of TLR signalling pathways [11]. scRNA-seq performed from ventral mesencephalic tissue obtained from postmortem PD patients and age-matched controls identified a disease-specific upregulation of microglia [12]. Interestingly, the authors identified a significant PD risk variant enrichment in microglia, showing the strongest association with the PD gene LRRK2 along with enrichment of NLRP3 inflammasome pathways [12]. In addition, a significant upregulation of GPNMB was found, which was associated with amoeboid microglia [12]. Of note, GPNMB is one of the most upregulated genes in DAM [13] and MGnD [9] phenotypes, and increased brain expression of GPNMB is associated with genome wide significant risk for PD [14].
A sustained and complex systemic activation of the immune system in PD is supported by increases of different cytokines (pro-inflammatory and anti-inflammatory) and immune-related molecules in CSF and serum of PD patients [15, 16]. From the different cytokines, TNF-α deserves special consideration as blocking TNF-α has been found neuroprotective in PD models [17] and usage of TNF-α antibodies has been found to lower PD incidence among patients with inflammatory bowel disease [18]. Increasing evidence supporting an important role of the peripheral immune system in PD pathology is evident and most of the peripheral contributors to PD-related neuroinflammation have been recently described by Romero-Ramos and colleagues [15]. Among them, monocytes/macrophages have been involved in PD pathogenesis since it is known that these cells infiltrate the brain during PD through the CCL2-CCR2 axis [19]. Furthermore, genetic profiling analysis has identified a distinct transcriptomic signature in monocytes from early PD patients [20]. Some researchers have also shown that monocytes from PD patients cannot produce a healthy and balanced response to different stimuli, such as α-syn for example [21]. The involvement of T cells in the pathogenesis of PD has been also demonstrated, since CD4+ and CD8 + T cells surrounding neuromelanin+ neurons have been detected in postmortem PD patients [22]. These observations pinpoint the interactions between brain innate and adaptive immune systems. The contribution of MHC II to PD pathology is inferred by studies demonstrating that MHC null mice are resistant to dopaminergic degeneration under conditions of α-syn overexpression [23]. Confirming these observations, genetic association with PD in the HLA region has been found, including HLA-DRA, HLA-DQA2, HLA-DQB1, HLA-DRB1, and HLA-DRB5 [24–27]. Importantly, a set of peptides derived from α-syn have been found to act as antigenic epitopes to further drive CD4+ and CD8+ cell responses in PD patients [28], thus linking preclinical studies and GWAS studies across the HLA regions. Finally, even though infiltrating B cells have not been detected in the brains of PD [22], a recent scRNA and BCR sequencing for B cells in PD patients and aged-matched controls identified increased memory B cells and increased IgG and IgA isotypes and more frequent class switch recombination events in PD patients [29]. All these findings have contributed to the redefinition of PD as a multisystemic disease that should be managed in a more integrative manner instead of the brain-focused classical approach. More research of this integrated network of communication that exists between peripheral immune cells and glial cells is necessary to improve our understanding of disease pathogenesis and hence provide more effective therapeutic approaches. All this information implies immune-associated models of PD as relevant tools to study the potential contribution of microglia to the degeneration of the nigrostriatal system and help in developing potential therapies for PD.
PD MODELS USING INTRACEREBRAL INJECTION
Lipopolysaccharide
Lipopolysaccharide (LPS; also known as endotoxin), a powerful pro-inflammogen, is the main component of the outer membrane of Gram-negative bacteria. The physiological response to LPS is mediated by the TLR4 in association with other proteins as the LPS binding protein (LBP), the monocyte antigen CD14, and the myeloid differentiation factor (MD)-2 [30]. Injection of LPS into different brain structures such as the cerebral cortex, striatum, choroid plexus-cerebral ventricles, or hippocampus triggers the activation of astroglia and microglia [31–34].
Since the death of dopaminergic neurons in SN is a key feature of PD, it was worth investigating whether injection of LPS into SN could induce a glial reaction and subsequent loss of dopaminergic neurons. A single intranigral injection of 2μg of LPS induces microglial activation and loss of astrocytes on the injection side, studied from two days after injection [35]. Using a single injection of 5μg of LPS into the SN, microglial activation has been described as early as 0.2 h after injection [36]. Importantly, the number of TH positive neurons on the SN is reduced on the ipsilateral side of injection [37]. Similarly, dopamine (DA) levels, its metabolites, and TH activity (a key enzyme in the synthesis of DA) decreased in both the SN and the striatum. The long-term analysis demonstrates that damage to the dopaminergic system is permanent, as seen one year after injection [35]. Other neuronal phenotypes, such as GABAergic or serotoninergic, are not affected, strongly suggesting that injection of LPS into the SN is a specific inflammatory animal model of PD [35]. Other authors have modified this model, using increasing amounts of LPS into the SN, as 5μg [36], 10μg [38], or up to 30μg [39]. Intrastriatal injections have also been used, employing either a low dose (0.05–5μg) [40] or a high dose (from 16 to 60μg) [41, 42]. Intrapallidal injection has also been reported (10μg) [43]. Interestingly, SN is always identified among the brain structures more prone to neuroinflammation. The causes of this situation have not yet been determined, although local differences in the number of microglia [44] and of the inflammation-related factors produced by these cells have been suggested [45]. In this sense, even systemic administration of LPS (which will be discussed in more detail below) has a specific effect on SN, increasing phagoptosis of nigral neurons through a mechanism dependent on the P2Y6 receptor [46].
Injection of LPS at a dose of 2μg did not affect the dopaminergic system when injected into the striatum or the medial forebrain bundle (MFB, the primary neural connection between these two structures) [35, 47]. However, it has been reported that the intrastriatal injection of 10μg of LPS in Sprague Dawley rats produces an inflammatory response, oxidative stress, and activation of the TLR/NF-KB (Nuclear factor-kappa B) pathway, with motor alterations [48]. Other authors administered even higher amounts of LPS (up to 60μg), inducing degeneration of the dopaminergic nigrostriatal system, motor impairment, and α-syn accumulation in nigral dopaminergic neurons [41, 42], with mitochondria affected before dopaminergic neuronal degeneration. Furthermore, a study using the injection of 10μg of LPS into the globus pallidus reported changes in SN iron levels, which could increase stress and subsequent vulnerability of nigral dopaminergic neurons [43]. It has been suggested that microglia could be responsible for differential susceptibility to LPS in brain structures [45]. On the other hand, models based on high LPS doses could deviate from physiological conditions not representing a ‘realistic’ disease model.
The effect of LPS on neuronal and glial cells is prevented by compounds with anti-inflammatory properties, such as dexamethasone (a potent and widely used anti-inflammatory drug) [49], minocycline (a tetracycline antibiotic) [50], simvastatin (a lipid-lowering agent) [51], or naloxone (an opioid receptor antagonist) [52]. It is striking that the LPS-induced neurotoxic effects in SN appear to be DA dependent since the inhibition of TH with α-methyl-p-tyrosine prevents microglial activation and LPS-induced damage to dopaminergic neurons [53]. Synergistic interaction of DA with other compounds to produce a toxic effect has previously been shown; of particular interest is the interaction with α-syn, which changes its aggregation pattern in vivo in contact with DA [54]. On the other hand, the contribution made by DA metabolism through monoamine oxidase (MAO, which produces H2O2) to oxidative stress should not be ruled out.
Stress reinforces the deleterious effect of LPS on SN. Therefore, the number of activated microglial cells in the SN of rats treated with LPS and the loss of astrocytes is almost doubled in stressed animals. The reinforcement by stress of the effect induced by LPS is similar (or even greater) on the expression levels of key proinflammatory molecules, including tumor necrosis factor (TNF), interleukin (IL)-1β, IL-6, and inducible NO synthase (iNOS), and the combined effect of stress and LPS results in a huge expression of monocyte chemoattractant protein 1 (MCP-1) mRNA. The number of TH positive neurons in the SN, which is halved in the animals treated with LPS, decreases to 25% of the control value when LPS is injected into the SN of stressed animals. RU486, a glucocorticoid receptor antagonist, prevents all these effects [55]. These data point to the potential role of stress in the initiation/development of the neurodegenerative process that leads to PD.
Thrombin
This multifunctional serine protease, well known for its participation in the blood coagulation cascade, has harmful effects on the CNS. When injected into the SN of Wistar rats, thrombin induces the expression of iNOS and proinflammatory cytokines (TNF, IL-1α, IL-1β) in both the SN and the striatum, increases microglial proliferation and activation, and induces the disappearance of astroglial cells around injection into the SN. Intranigral injection of thrombin also reduces the number of dopaminergic neurons in this structure without affecting other neuronal phenotypes such as GABAergic neurons. Similar results were described in Sprague Dawley rats [56], including the activation of apoptosis and the c-Jun N-terminal kinase (JNK) and p53 signaling pathways [57]. When injected into the striatum, thrombin induces a retrograde loss of dopaminergic neurons in the SN, also affecting the fibers immunopositive for TH that connect both structures and inducing the formation of deposits of α-syn in the SN, a hallmark of PD [58]. Blocking PAR4, a thrombin receptor, prevents these effects suggesting that thrombin could be involved in eliminating presynaptic elements in the striatum, leading to synaptic loss [59].
Anti-inflammatory compounds prevent the effect of compounds that trigger an inflammatory response. For example, minocycline-induced suppression of reactive oxygen species (ROS) derived from NADPH oxidase and expression of proinflammatory cytokines prevented the death of thrombin-induced dopaminergic neurons induced by thrombin [60]. However, in the intranigral thrombin model, systemic administration of dexamethasone, a widely used anti-inflammatory drug, does not only fail to prevent microglial activation but increases dopaminergic neuron damage. In fact, dexamethasone does not decrease the number of apoptotic cells, nor reduces thrombin-induced α-syn deposits, but reduces the amount of P-Akt. Interestingly, these effects appeared to be mediated by increases induced by thrombin in MAO, which was prevented by the MAO inhibitor tranylcypromine [61]. This suggests that in cases in which the integrity of the blood-brain barrier (BBB) has been compromised and thrombin is in contact with the cerebral parenchyma (as occurs in processes that affect the cerebral vasculature, such as stroke), the administration of dexamethasone as an anti-inflammatory therapy would be counterproductive.
α-Synuclein fibrils injections
α-syn protein injection has been widely used in the last decade to promote PD-like features focused on α-syn aggregation. In particular, striatal injection of α-syn pre-formed fibrils (PFF) demonstrated to cause Lewy body-like inclusion and dopaminergic degeneration in mice [62]. The relevance of neuroinflammation in this model was recently investigated in mice by Earls et al. [63] demonstrating upregulation of MHC-II as a signal of microglia activation while also describing astrogliosis and lymphocyte infiltration while similar results were obtained in rats [64]. Interestingly, T-lymphocytes have been proposed to limit phosphorylation of α-syn [65]. Injection of PFFs in transgenic models also demonstrated the importance of neuroinflammation. For instance, injection of PFF in A30P transgenic mice also resulted in increased microgliosis [66]. Similarly, injection of PFF in A53T transgenic mice led to microglia activation and neurodegeneration. However, genetic deletion of TLR2 or pharmacological inhibition achieved to decrease microgliosis and cytokine release while also protecting the dopaminergic system [67]. Overall, injection of PFF is, nowadays, one of the most used models for the study of α-syn aggregation and transmission presenting interesting similarities with the progression of PD in humans. However, the inflammatory component of this model is very relevant and should be considered as one of the best choices for determining the role of neuroinflammation in PD progression.
Other models using intracerebral injection
The tissue-type plasminogen activator (tPA, another serine protease) was the first drug approved (1995) by the Food and Drug Administration to treat acute ischemic stroke. However, beyond its beneficial abilities as a clot-dissolving agent, injection of tPA into Wistar rats’ SN produces microglial activation and loss of astrocytes, degeneration of dopaminergic neurons without affecting GABAergic, disruption of BBB, α-syn deposits, increased expression of the brain-derived neurotrophic factor (BDNF), nNOS and iNOS, and alteration of phosphorylation levels in the proteins JNK, p38, extracellular signal-regulated kinases (ERK), Akt, glycogen synthase kinase (GSK)-3β and cAMP responsive element-binding protein (CREB) [68].
Trisialoganglioside (GT1b; a glycosphingolipid containing sialic acid) is a surface molecule of mammalian cells with endogenous effects on the CNS. Injection of GT1b into the SN of female Sprague-Dawley rats induced the loss of NeuN and TH positive neurons in this structure in a dose-dependent manner. GT1b induced microglial activation and expression of iNOS in microglia (as soon as 4 h after injection). Inhibition of NOS by L-NG-nitroarginine methyl ester (L-NAME) partially prevented the deleterious effect of GT1b [69].
Finally, neuromelanin is a pigment found in human catecholaminergic neurons; however, extracellular neuromelanin has been suggested to activate microglial cells [70]. Zecca et al. [71] published a new model of microglial activation and 50% of dopaminergic degeneration after intranigral injection of human neuromelanin in rats.
Virus-mediated overexpression of proteins
Overexpression of α-syn has been used in the last decade as a model of parkinsonism focused on the aggregation capacities of α-syn inside the dopaminergic neurons [72]. Different adenovirus-associated vector (AAV) serotypes have been used to induce the expression of wild-type or mutant human α-syn. This overexpression is frequently associated with a neuronal promoter and locally injected in the midbrain region to investigate the effect on dopaminergic neurons (see [73]). Inflammation has been closely related to this model, for instance, Sanchez-Guajardo et al. [74] first described early microglia activation in rats midbrain after AAV2/5 serotypes injections along with lymphocytes infiltration. Same serotype was demonstrated to induce microglia activation independently of neurodegeneration in monkeys [75] and mice [76]. Neuroinflammation appeared in the striatum even before than in the SN, including increased levels of several cytokines like IL-1β and TNF-α [77]. Furthermore, inhibition of microglia activation has been demonstrated to protect dopaminergic integrity after AAV9 serotype injection [78] and AAV2 serotype where MHC-II genetic deletion resulted in absence of neurodegeneration [23]. Conversely, further activation of microglia through LPS injection promoted cell-to-cell transmission of α-syn [79]. Interestingly, combination of this model with injection of PFF has also shown to increase microglia activation, microgliosis and dopaminergic degeneration [80].
However, Bido et al. [81] have recently published a novel variant of this model focusing on the effect of α-syn on microglial cells. In their study, they used a novel lentiviral FLEX system of conditional gene expression to provoke microglia-specific overexpression of mutant A53T α-syn associated with the expression of CX3CR1 receptor. The authors achieved high cell specificity with this method and discovered that microglial A53T α-syn overexpression promoted microglial activation and dopaminergic degeneration. Surprisingly, no intraneuronal α-syn accumulation was found, but rather microglia presented signs of α-syn accumulation like phosphorylation in serine S129. Microglia has been proposed as the cell responsible for pathological α-syn degradation. In fact, Heneka and colleagues have recently demonstrated that microglial cells can transport pathological α-syn from microglia to microglia through tunneling nanotubes for cooperative degradation [82]. However, under these conditions, α-syn fibrils induced the production of ROS, resulting in a compromised plasma membrane and mitochondrial network disintegration [82]. Both studies highlight the ability of microglia to isolate and degrade pathological α-syn. However, it is important to keep in mind that levels of pathological α-syn rely on the perfect balance of three independent processes associated with α-syn homeostasis: formation, aggregation rate, and clearance [83]. This view is exemplified in the model used by Bido et al. associated with overexpression of α-syn that provoked microglial exhaustion, inefficient degradation, microglia activation, and neuronal degeneration. Importantly, under these conditions, microglia showed a transcriptomic profile with upregulation of main proinflammatory cytokines like Il1b and Tnfa as well as several chemokines. Additionally, authors discovered upregulation of some of the genes related to DAM phenotypes discovered in other neurodegenerative diseases models like Apoe or Itgax [8, 9]. Consequently, any disturbance of such a balance may make microglia prone to produce high levels of neurotoxic ROS and proinflammatory factors, ultimately leading to cell death. Similarly, Zhang and colleagues also promoted α-syn overexpression in microglia primary cultures and microglial cell lines [84], promoting a robust inflammatory response and cytokine release that could be impaired by the mGluR5 receptor activation.
While the recent study from Bido et al. has shed light on the implication of α-syn in microglia activation and consequent dopaminergic degeneration, other studies had previously used viral-mediated overexpression to promote microglial activation. For instance, in 2006, Ferrari and colleagues [85] proved that chronic overexpression of IL-1β in the SN leads to progressive neurodegeneration in rats associated with microglial activation. Similarly, overexpression of TNF led to mild but progressive neurodegeneration as soon as 14 days [86]. Notably, both studies suggested independent effects of both cytokines, as levels of IL-1β after TNF overexpression remained low and vice versa.
Interestingly, the combination of adenoviral expression with classical PD models could become a novel strategy for studying different immunomodulatory proteins and deciphering PD-specific microglial phenotype. For instance, Ren et al. have studied the role of TREM2 overexpression in a model of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). TREM2 is thought to be a master regulator of microglia phenotype. Upon activation, TREM2 promotes phagocytosis, ameliorated inflammatory response and neuroprotection, but is also a key receptor for neurodegenerative microglia [9] and is needed for complete inflammatory response (reviewed in [87]). In the context of dopaminergic degeneration, overexpression of TREM2 led to decreased neuroinflammation and dopaminergic protection [88], which goes in line with later studies focused on TREM2 knockout mice and supports the view that the DAM phenotype is neuroprotective [89].
Altogether, adenoviral overexpression offers a broad panel of possibilities for studying microglial activation as a PD model and represents a novel tool that should highly impact future PD research.
The experimental animal models of PD described so far, in one way or another, end up activating this inflammatory response, indicating that the different models of this disease show common mechanisms to some extent.
As shown above, LPS is a direct inflammatory model. On the contrary, substances such as 6-OHDA MPTP/MPP+, paraquat, or rotenone are classic toxic models that produce the specific death of dopaminergic neurons. Interestingly, its administration eventually triggers a harmful inflammatory reaction. In recent years, the idea that neuronal death produced by these toxic substances was followed by activation of the immune response has changed towards a scenario in which the latter is activated before (or even in the absence of) neuronal death, so that microgliosis and not neurotoxicity would be the determining factor in neuronal death. For example, MPTP administration not only induced microglial activation but also induced T lymphocyte (CD4+ and CD8+) infiltration into the brain of non-human primates [90]. Thus, both peripheral and cerebral immune responses are involved in the mechanism of death induced by MPTP.
6-OHDA induces a strong production of free radicals that was soon identified [91, 92], in addition to inhibition of complex I of the mitochondrial respiratory chain, a mechanism shared by substances such as MPTP/MPP+and pesticides such as rotenone and paraquat. In the 6-OHDA model, the loss of astrocytes and the alteration of the BBB, the death of dopaminergic neurons, and the activation of microglia observed in the SN and the striatum was accompanied by the infiltration of peripheral immune cells [93]. Interestingly, these effects are prevented in female TLR4 KO animals (suggesting a gender-dependent mechanism) [94], and the treatment with urocortin [95] or P2Y6R KO [46] exert protection. Since these substances also exert protection in the LPS model, the inflammatory response arises as a common mechanism to different molecular challenges showing that the inflammatory response induced by LPS and 6-OHDA share common pathways.
The effect of α-syn overexpression in dopaminergic neurons of the SN produces a down-regulation of TH and an increased sensitivity to MPTP/MPP+ [96]. Interestingly, α-syn can also exert a detrimental effect on mitochondria, altering complex I-dependent respiration [96, 97]. α-syn exerts an unquestionable activating effect on microglia, especially the misfolded forms [98], which extends to monocytes [99, 100]. The Lewy pathology and neuroinflammation can then mutually potentiate in a vicious cycle, facilitating the progression of the pathology and the death of dopaminergic neurons [101, 102].
The involvement of the peripheral immune cells in these PD models has also been described. For instance, peripheral immune components infiltrate the brain following intracranial injection of any of the toxic mediators discussed. Injection of LPS into the SN causes infiltration of peripheral macrophages, which contributes to the observed damage; in fact, the depletion of peripheral macrophages using clodronate not only eliminates their infiltration, but also reduces other harmful effects of LPS injection, such as microglial activation, loss of astrocytes, disruption of the BBB, and death of dopaminergic neurons in the SN [103]. In the 6-OHDA model, the loss of astrocytes and the alteration of the BBB, as well as the activation of microglia and the infiltration of peripheral immune cells observed in the SN and the striatum, decreased when the concentration of DA was depleted by the TH inhibitor α-MPT, suggesting an interaction between endogenous DA and toxins [93].
Any condition affecting the integrity of the BBB can potentially let (or maybe induce) the infiltration of peripheral immune cells. Alteration of the BBB permeability has been shown in several animal models of PD, as for the intranigral/intrastriatal injection of thrombin [57] or tPA [68].
Circulating neutrophils, for example, are important in ischemic stroke [104, 105], where they become the main producers of matrix metalloproteinases (MMP-9), disruptors of the BBB. Peripheral immune cells are arising as interesting therapeutic targets in brain disorders coursing with inflammation; thus, the treatment with L-cysteine (a source of SH2 groups) reduced infiltration of peripheral immune cells in the brain, contributing to a better outcome of neuronal deficits induced by LPS [106]. Overexpression of α-syn in microglial cells induced by lentivirus produces an inflammatory cycle involving infiltrating immune cells [81].
PD MODELS COMBINING PERIPHERAL AND CENTRAL INFLAMMATION
The data discussed above make clear the pivotal role of neuroinflammation in the development of PD. Nevertheless, the increase in understanding of PD has led Brundin and colleagues to redefine its pathogenesis by dividing the course of the disease into three temporal phases mediated by triggers, facilitators, and aggravators [107]. Following this concept, the inflammatory models described so far could be the trigger that initiates the neurodegenerative process. However, in the context of PD, triggers alone may be insufficient for the pathology of PD to develop, requiring facilitators. Consistent with this view, our group was a pioneer in pointing to peripheral inflammation as one of these facilitators. In 2010, Villarán et al. described that peripheral inflammation induced by a model of ulcerative colitis based on the administration of dextran sulfate sodium (DSS) in drinking water exacerbates LPS-induced damage to the nigral dopaminergic system [103]. The contribution of chronic peripheral inflammation to the pathogenesis of neurodegenerative diseases is now an outstanding question. In the past 10 years, several clinical data and animal models have supported this view, suggesting peripheral inflammation as a potential risk factor in neurodegenerative diseases, especially in PD (for a review, see [108]). Sustained activation of the peripheral innate and adaptive immune systems occurs in the context of a wide range of disorders ranging from chronic infectious diseases to autoimmune and metabolic diseases, such as obesity, diabetes mellitus, and atherosclerosis. In addition, it is increasingly recognized that progressive systemic inflammation takes place during aging, a term known as inflammaging. Chronic peripheral inflammation that accompanies these diseases has been proposed to induce the production of proinflammatory cytokines that, following the endocrine route or through the vagus nerve transmission, can enter the brain [109]. In addition, increasing levels of proinflammatory cytokines compromise the permeability of the BBB, allowing many immune blood cells, including monocytes and T cells [110], to cross the altered BBB. This realization arises from multiple clinical studies showing elevated levels of inflammatory mediators in patients with PD, providing strong evidence for the interplay of the innate and adaptive immune system in the CNS and periphery in the context of PD and other synucleinopathies [15]. Some authors have proposed that this humoral immune response could be correlated with the nonmotor symptoms of PD [109]. In this context, we can face two possible scenarios: peripheral inflammation can “prime” microglial cells, which may become over-activated when a second noxious stimulus arrives. On the other hand, peripheral inflammation can transform previously “primed” microglia into an activated state. In both cases, peripheral inflammation can trigger stronger responses and further perpetuate the ongoing neurodegenerative process [111, 112].
Take-home information. All the models described so far share two common features: microglial activation and the death of dopaminergic neurons. This can be achieved by central or peripheral inflammatory challenges while their combination leads to a higher effect
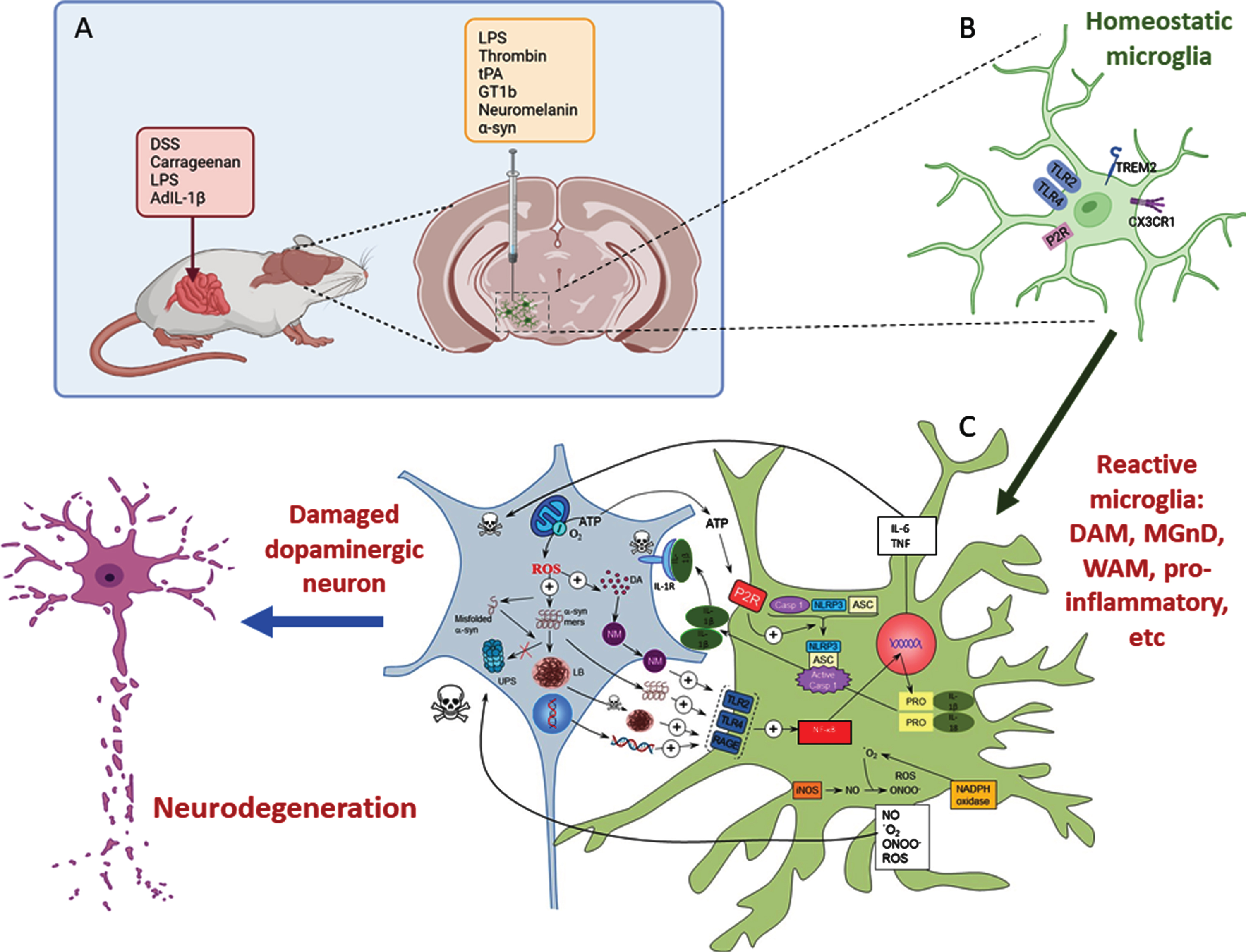
A) Inflammatory models of Parkinson’s disease take advantage of the use of different proinflammatory compounds administered peripherally, intracerebral, or by combining both pathways. B) Whatever compound and route of administration used, homeostatic microglia sense the environment through a set of surface receptors (the pattern recognition receptors, PRRs), including TLR2, TLR4, and RAGE. C) When activated, microglia undergo molecular and morphological changes, becoming reactive microglia. Illustrative examples are the DAM phenotype driven by TREM2 or the proinflammatory phenotype driven by TLR activation. Different microglia phenotypes can coexist under neurodegenerative conditions. Their activation leads to activation of the NF-κB pathway and the transcription of several proinflammatory genes (TNF and IL-6). Assembly of the NLRP3 inflammasome and activation of caspase-1 produce IL-1β and IL-18. Reactive microglia are also a source of ROS and RNS. All these products exert a harmful effect on dopaminergic neurons, which in turn release substances such as ATP, neuromelanin, and different forms of α-syn (either monomers or aggregates) that bind microglial PRRs in a vicious cycle that eventually leads to the death of dopaminergic neurons. Modified from Herrera et al., 2018 [169] using BioRender.
Considering all this information, some authors, including our group, have developed several animal models resulting from the combination of peripheral inflammation and central nigral dopaminergic challenge (reviews in [113, 114]). These models have been summarized in Table 1.
Models based on the combination of central and peripheral inflammation used to study the implications of systemic inflammation in the development of PD
The most popular model of peripheral inflammation used in these combined models is the one based on intraperitoneal injection of LPS at doses ranging from 0.4 to 2.5 mg/kg in just one or several consecutive days. Gut inflammation induced by the ulcerative colitis model based on DSS administration (from 0.5 to 5%) in drinking water is the other most common model of peripheral inflammation. Injection of carrageenan into the paws of rats and injections of adenoviral vector that produce human IL-1β are also useful approaches to achieve peripheral inflammation. These models of peripheral inflammation were combined with a nigral insult induced by several PD models, including the 6-OHDA, MPTP, LPS, rotenone, and paraquat administration models. Inhibition of the proteasome system and injections of α-syn oligomers or transgenic animals that overexpress α-syn are also used as PD models. To note, chronic stress, which is a common condition nowadays, can influence the gut microbiota and alter the complex equilibrium in the intestinal milieu leading to a proinflammatory state that has been shown to accelerate neuronal degeneration and motor deficits in parkinsonism rodent models [124].
All these data reinforce the idea that peripheral inflammation could be a significant risk factor for PD and, therefore, strategies aimed at controlling the systemic inflammatory state arise as potential therapeutic options to control the development of PD. In this sense, Boza-Serrano et al. have shown that modulation of galectin-3, a microglia-related protein recently described as an immunomodulator, plays a significant role in microglia activation induced by α-syn [128]. Indeed, we have demonstrated that this ability of galectin-3 is extensive to emerge as a promising strategy to minimize undesired microglia activation states in PD [129]. The use of anti-inflammatory therapies in PD treatment has also been proposed, although clinical trials do not show significant results [130]. The NLRP3 inflammasome plays a critical role in the pathogenesis of PD, which led some authors to propose liver NLRP3 inhibitors to attenuate systemic inflammation and protect against a model of PD in rodents [131]. Finally, Liu et al. have demonstrated that peripheral immune tolerance mediated by CD200/CD200R signaling can attenuate neuroinflammation and decrease neurodegeneration in the LPS model of PD, suggesting CD200R as a potential therapeutic target to alleviate neuroinflammation in PD [132, 133].
MODELS OF PD BASED ON PERIPHERAL INFLAMMATION
All these studies target systemic inflammation as a possible facilitator of PD. The question is whether peripheral inflammation is able per se and without any other central stimulus to induce a neuroinflammatory environment in the brain that subsequently could induce dopaminergic neurodegeneration. In this sense, two important PD models based on peripheral inflammation are arising: the systemic LPS injection and the gut-brain axis.
Systemic LPS injection
The relevance of systemic inflammation in the integrity of the dopaminergic system was first demonstrated by Qin et al. in 2007 [134] when they administered a single dose of intraperitoneal LPS (5 mg/kg) in adult mice and discovered that the mice suffer from chronic and progressive dopaminergic degeneration at 7 and 10 months after the injection. Interestingly, LPS is not reported to cross the BBB, but authors identified the upregulation of peripheral TNF as the responsible for dopaminergic vulnerability. Indeed, effects of injection include increased microglial activation and reduced TH staining in the first hours post-injection [135]. Notably, the study by Qin et al. stimulated the combination of systemic LPS injection with other parkinsonian models (see Table 1). Later, other studies have observed a more accelerated degeneration with repeated injections of LPS. For instance, Bodea and colleagues [136] proposed that systemic injection of LPS during 4 consecutive days (1 mg/kg per day) led to dopaminergic degeneration 15 days after the last injection, in contrast with the same dose in a single injection that was unable to promote degeneration at that time point. Systemic LPS injection was characterized by an increased initial microglial response with significant cytokine production, particularly TNF and IL-1β; however, this activation returned to basal levels 15 days after the injection. Importantly, systemic LPS can be a valuable model for studying prodromal PD. For instance, systemic LPS models present early α-syn alterations and non-motor symptoms in the gut [137], olfactory impairments, and anxiety-like behavior [138]. Indeed, Song and colleagues [139] demonstrated that systemic LPS promotes sequential degeneration in the brain, resembling the initial phases of the Braak theory [140], starting in the locus coeruleus, followed by the SN, and lastly, the cortex and hippocampus.
An innovative variant of the effect of peripheral LPS on the dopaminergic system is the chronic intranasal administration, which after 5 months of daily administration, promoted microglia activation, moderate (∼50%) dopaminergic degeneration, and, remarkably, α-syn aggregation [141]. A similar but shorter model was also used by Li et al. [142], leading to a 38% of TH neuronal loss after 1 month of treatment, increased α-syn expression, and behavioral impairment. The same approach was used by Niu et al. [143], describing a more intense nigral degeneration after 6 weeks of treatment while also identifying IL-1β signaling as a relevant factor in this model.
Gut-brain axis
Different authors have recently suggested that intestinal inflammation could be a silent driver of PD pathogenesis [144]. The term gut-brain axis has been progressively gaining interest in the last 20 years. This term refers to the bidirectional communication between the CNS and the enteric nervous system and incorporates the fine regulation of immune responses in the gut and brain [144]. It is known that inflammatory processes can enter the CNS through different mechanisms, including the humoral and neuronal pathways (see [113]). Braak and colleagues, based on the appearance of Lewy pathology, already hypothesized the possibility that PD may start in the gastrointestinal tract to spread to the brain via the vagus nerve to further reach the ventral mesencephalon [140]. Indeed, experimental evidence has shown that the gastrointestinal tract is a potential starting point for aggregated α-syn, with the vagus nerve acting as a route by which pathology may be transmitted to the lower brainstem [145]. Therefore, a new model for PD pathogenesis has been recently proposed [144]. In this model, the disorder originates in the intestine to further progress to the ventral mesencephalon in an inflammation-mediated process. Thus, in a susceptible individual, inflammatory triggers, such as bacteria, viruses, or environmental toxins could initiate immune responses in the gut that eventually could deleteriously impact the microbiota, increasing intestinal permeability and inducing increased expression and aggregation of α-syn. Aberrant conformations of α-syn may be transmitted from the gut to the brain via the vagus nerve, while chronic intestinal inflammation promotes systemic inflammation. As mentioned before, this peripheral inflammation can increase BBB permeability, allowing the entrance of cytokines and immune blood cells to the brain parenchyma. Combination of intestinal inflammation, systemic inflammation and α-syn pathology in the brain promote neuroinflammation, which eventually drives the neurodegeneration process that characterizes PD. This model is sustained by epidemiological data showing that patients with inflammatory bowel disease (IBD) have a higher risk of developing PD than non-IBD individuals [146]. Moreover, gene association studies have found a genetic link between PD and IBD [147]. Therefore, it would be interesting to look for parkinsonian signs in animals using models to mimic IBD pathology. In this context, Labandeira-García’s group showed that a subchronic regimen of 2.5% of DSS for three weeks results in early changes in the nigrostriatal dopaminergic homeostasis, dopaminergic neuronal death, and increased levels of nigral proinflammatory mediators [148]. These are intriguing data that deserve further investigation since, if confirmed, this model would greatly contribute to understanding the underlying mechanisms involved in PD.
Anti-inflammatory interventions
All this information has encouraged some authors to deepen their understanding of the effects of peripheral inflammation on neuroinflammation. These studies have revealed that peripheral inflammation, especially gut inflammation, induces neuroinflammation in certain brain structures that is accompanied by several manifestations such as anxiety, depression, chronic pain and memory and cognitive impairments [149, 150]. However, this neuroinflammation and its associated symptoms decrease with some anti-inflammatory interventions in animal models. These treatments include inhibitors of the S-100 protein, TNF inhibitors, and neutrophil depletion [151, 152]. Melatonin, fermented rice brand, and DHA/EPA treatments also improve the symptoms associated to peripheral inflammation-related neuroinflammation [153–155]. In this regard, our group has recently published a study on the peripheral and central anti-inflammatory effects of galectin-3 inhibitors in DSS-induced gut inflammation [129].
There is, therefore, increasing interest in testing these anti-inflammatory treatments in humans, which is why several clinical trials are running (CN-02323358). Considering that intestinal inflammation appears to be the most powerful driver of neuroinflammation, most of these trials focus on the reduction of gut inflammation, modifying the microbiota through probiotics and prebiotics (CN-02355534; NCT04512599; NCT05146921; NCT04032262, NCT05173701, NCT04159727). However, to date, only one trial has been completed [156]. In this study, the authors evaluate the effects of probiotic supplementation on inflammation-related gene expression in PD patients, finding an overall significant improvement on several inflammatory-related genes such as IL-1, IL-8, TNF-α and TGF-β. Further studies need to be completed to gain a better understanding of whether interruption in inflammatory signaling ameliorates inflammation and subsequent neurodegeneration.
Viral parkinsonism models
A viral onset has been long proposed for PD since 1918 influenza led to some encephalitis cases that mimicked some parkinsonism symptoms [157]. It remains however unclear if there is a real implication of viral infections on PD onset [158]. However, viral infection can lead to systemic hyperinflammation known as “cytokine storm” that can penetrate in the brain and promote a potent inflammatory response, oxidative stress and upregulation of α-syn [159, 160]. In Sadasivan et al. [161], authors examined the effect of H1N1 influenza virus in an MPTP model of PD. The authors observed an increased loss of dopaminergic neurons but they failed to attribute it to increased microglia activation, what suggests an implication of peripheral immune system, but also the direct action of the virus in the dopaminergic neurons [162]. However, the best described virus used to induce PD in animal models has been Japanese encephalitis virus [163]. In this model, strong microglia reactivity has been observed through TLRs activation while also compromising dopaminergic system integrity [164]. Similarly, alphaviruses have also been described as an alternative parkinsonism model [165].
The new pandemic caused by coronavirus SARS-CoV-2 has gained a lot of attention from a PD research perspective. Despite the relation of COVID-19 with PD, this is yet not clear as the neurological effects of COVID-19 are still arising [166]. It has been reported that PD patients could develop prolonged post-COVID19 syndrome with worsen motor behavior and poor levodopa response [167]. Several COVID-19 features could lead to worsening PD symptoms; in particular, those related with cytokine storm that increase serum levels of distinct cytokines and neurotoxic components that have been previously related with PD [168]. No model for the impact of SARS-CoV-2 in PD, or vice versa, has been proposed. However, health care systems overload, pandemic-derived psychological stress and lockdown restrictions have had a major (but variable) impact on PD patients’ status that should be carefully addressed before analyzing any physiological effect of COVID-19 on PD etiology.
CONCLUSIONS
This plethora of immune models may help to understand the complex molecular mechanisms associated with PD like the contribution of central and peripheral immune cells in key events of the disease. Among them, we may cite the role in the aggregation and spreading of α-syn and identification of microglia subtypes and their contribution in the disease. Elucidation of signaling pathways behind these events may be critical for identification of preclinical drugs potentially relevant for PD.
Footnotes
ACKNOWLEDGMENTS
This work was supported by grants from the Spanish Ministerio de Ciencia, Innovación y Universidades (RTI 2018-098830-B-I00), from the Consejería de Economía y Conocimiento of Junta de Andalucía (P18-RT-1372 and US-1264806).
CONFLICT OF INTEREST
The authors have no conflict of interest to report.