
Abstract
Background:
The concept of motor reserve explains the individual differences in motor deficits despite similar degrees of nigrostriatal dopamine depletion in Parkinson’s disease (PD).
Objective:
To investigate glucocerebrosidase (GBA) variants as potential determinants of motor reserve for exploratory purposes.
Methods:
A total of 408 patients with drug-naïve PD were enrolled from the Parkinson’s Progression Markers Initiative cohort database. All patients underwent SPECT dopamine transporter (DAT) scans and had results for Sanger sequencing of GBA. Parkinsonian motor deficits were assessed using the Movement Disorders Society Unified Parkinson’s Disease Rating Scale Part III (MDS-UPDRS-III). We compared MDS-UPDRS-III scores while adjusting for DAT availability in the putamen (i.e., motor reserve) between the PD groups according to the presence of GBA mutations.
Results:
Fifty-four (13.2%) patients carried GBA mutations. PD patients with GBA mutations were younger than those without mutations. There were no significant differences in sex, disease duration, years of education, and striatal DAT availability between the PD groups. PD patients with GBA mutations had higher MDS-UPDRS-III scores for the less affected side than those without mutations, despite similar levels of DAT availability in the contralateral putamen. The MDS-UPDRS-III sub-scores of the more affected side did not differ between the two PD groups.
Conclusion:
The results of this study demonstrated the detrimental effect of GBA variants on individual capacity to cope with PD-related pathologies, with different impacts depending on the motor laterality.
Keywords
INTRODUCTION
The concept of reserve was introduced to explain the individual variability in clinical manifestations despite the similar degree of pathological brain changes in neurodegenerative disorders [1]. The most representative form of reserve is the cognitive reserve in Alzheimer’s disease (AD) [2], while the concept of motor reserve in Parkinson’s disease (PD) is also increasingly accepted to explain the mismatch between parkinsonian motor deficits and the degree of nigrostriatal dopamine depletion in individuals with PD [3]. Our previous works demonstrated that educational experience [4], premorbid exercise engagement [5], and dominant-side laterality [6] could enhance the motor reserve in patients with newly diagnosed PD, providing resilience in the face of substantial PD-related pathology [7]. We also identified the functional brain network associated with motor reserve, comprising the basal ganglia, hippocampus, amygdala, inferior frontal cortex, insula, and cerebellar vermis [8]. As the clinical heterogeneity of PD may reflect different genetic backgrounds [9], some genetic variants could determine the motor reserve in individuals with PD. While some reports have suggested genetic factors related to cognitive reserve or resilience to AD-related pathology [10, 11], few studies have investigated the link between genetic factors and motor reserve in PD populations.
Mutations in the glucocerebrosidase (GBA) gene are one of the most common genetic risk factors for PD. Although the exact mechanisms underlying the increased risk of PD in patients with GBA mutations are not fully understood, several explanations have been proposed, including lysosomal dysfunction, increased endoplasmic reticulum stress, and mitochondrial impairment [12]. Moreover, recent evidence has suggested that genetic variation in GBA strongly affects the natural history of PD, including more rapid motor progression and cognitive decline [13, 14], as well as greater non-motor symptom burden [15]. Based on these findings, we hypothesized that GBA variants would have detrimental effects on individual capacity to cope with PD-related pathologies. Thus, this study investigated whether GBA mutations could be a determinant of motor reserve in patients with newly diagnosed PD for exploratory purposes.
METHODS
Subjects
We used the database from the Parkinson’s Progression Markers Initiative (PPMI) cohort, an observational, international study cohort designed to identify clinical, imaging, genetic, and biospecimen PD progression markers to accelerate disease-modifying therapeutic trials [16]. We downloaded clinical, imaging, and genetic data of newly diagnosed drug-naïve patients with PD from the PPMI database in April 2021. A total of 408 drug-naïve and untreated patients with PD, who underwent dopamine transporter (DAT) imaging using 123I-Ioflupane single-photon emission computed tomography (SPECT) with results for Sanger sequencing of GBA, were enrolled in this study (Fig. 1). Movement Disorders Society Unified Parkinson’s Disease Rating Scale Part III (MDS-UPDRS-III) was used to assess the severity of motor deficits in each patient at the time of DAT scan acquisition [17]. Cognitive function was assessed using the Montreal Cognitive Assessment (MoCA) and a standardized cognitive assessment battery which covers four cognitive domains (letter number sequencing for attention and working memory; semantic fluency and symbol digit modalities test for executive function; Hopkins Verbal Learning Test (HVLT) for memory; and Benton’s Judgement of Line Orientation for visuospatial function) [18]. Additionally, the sum of the scores of MDS-UPDRS-III items (rigidity, finger tapping, hand movements, pronation-supination movements of hands, toe tapping, leg agility, postural tremor of the hands, kinetic tremor of the hands, and rest tremor amplitude) was calculated for each side of the body (i.e., more affected and less affected sides).
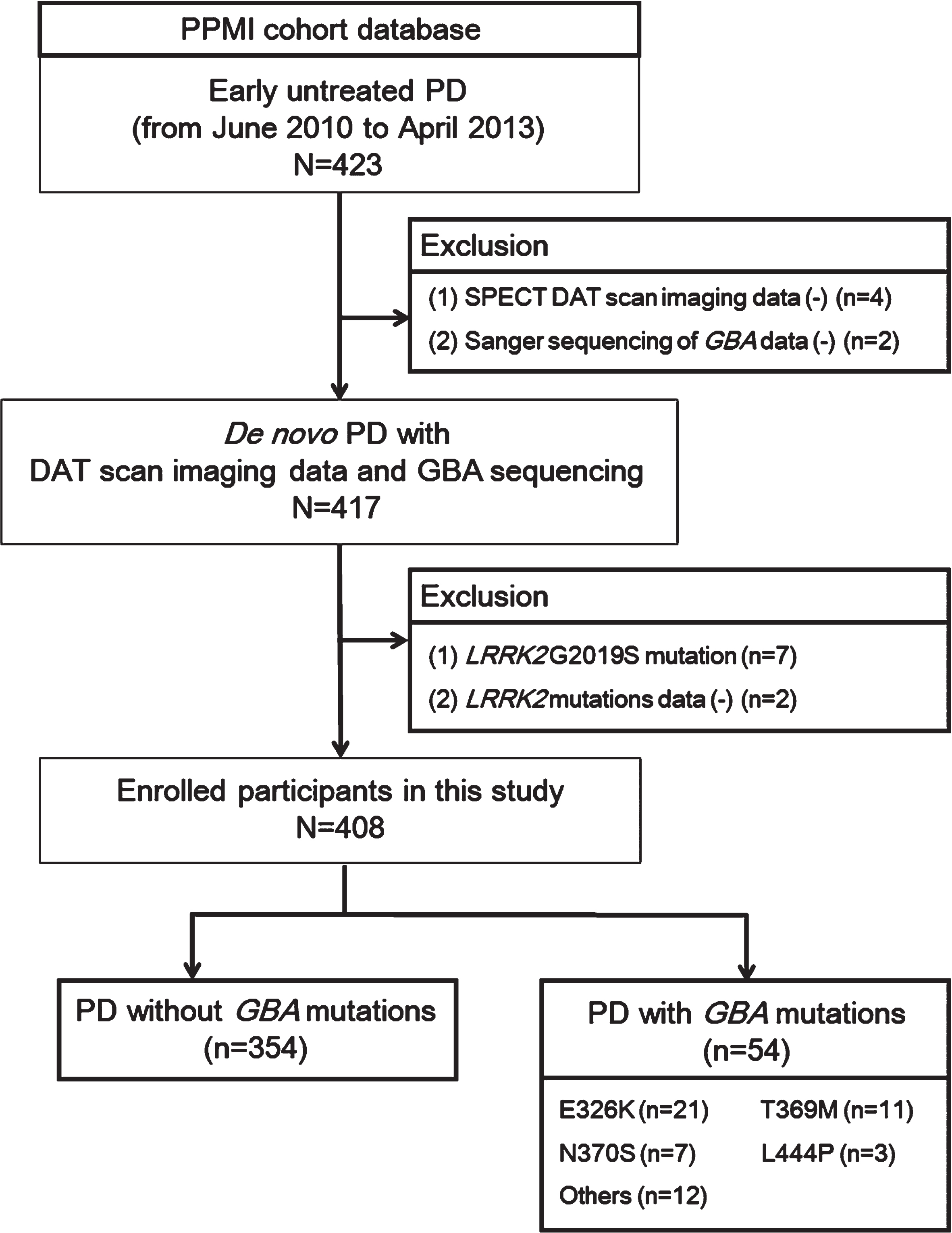
A flowchart of participants and enrollment.
Standard protocol approvals, registration, and consent
The study was approved by the Institutional Review Boards of each participating PPMI site. Written informed consent was obtained from all participants. The study is registered at http://www.clinicaltrials.gov (identifier: NCT01141023).
Quantitative analyses of SPECT DAT scan imaging data
SPECT DAT scan imaging data were acquired at PPMI imaging centers per the PPMI imaging protocol and sent to the Institute for Neurodegenerative Disorders for processing and calculation of striatal binding ratios (SBRs) [19]. The regions of interest were placed on the left and right caudate, left and right putamen, and occipital cortex (reference region). The SBRs for each of the four striatal regions were then calculated as (target region/reference region) –1. The mean of SBRs in the bilateral putamen was also calculated to estimate the motor reserve as DAT availability in this region is closely associated with the severity of PD motor symptoms [20].
GBA genetic testing in the PPMI
Sanger sequencing was performed on whole-blood extracted DNA samples collected according to the PPMI Research Biomarkers Laboratory Manual [18]. Exons 1–11 within GBA were Sanger sequenced and screened for variants. Patients who had or were not tested for the leucine rich kinase 2 G2019S mutation were excluded from this study (n = 9).
Statistical analyses
The baseline demographic characteristics and striatal DAT availability were compared between the PD groups according to the presence of GBA mutations using Student’s t- and Pearson’s χ2 tests for continuous and categorical variables, respectively. MDS-UPDRS-III scores were compared between the PD groups by analysis of covariance (ANCOVA) while adjusting for age, sex, disease duration (i.e., time from symptom onset to diagnosis), and the natural logarithm of SBR in the putamen as covariates. MDS-UPDRS-III sub-scores of the more affected and less affected sides were also compared by ANCOVA while adjusting for age, sex, disease duration, and the natural logarithm of SBR in the contralateral putamen.
Additionally, the motor reserve of each patient was estimated using the residual-based approach, as described in our previous works [8, 21]. In detail, we used the general linear model to predict the MDS-UPDRS-III score by using age, sex, disease duration, and the natural logarithm of SBR in the putamen (Supplementary Table 1). Then, the residuals (i.e., differences between the actual and predicted MDS-UPDRS-III scores) in the general linear model were calculated and standardized. A greater standardized residual indicated that the subject had higher MDS-UPDRS-III score than predicted (i.e., lower motor reserve). We defined the “motor reserve estimate” of each patient as a negative value of standardized residual in the general linear model, with high values indicating high motor reserve. A similar approach has been applied in some previous studies [22, 23], where the residual model was used to define the cognitive reserve in AD populations. The proportions of patients with low motor reserve estimates and high motor reserve estimates (cut-off, 0) were compared between the PD groups using Pearson’s χ2 tests. The same analyses were applied to the MDS-UPDRS-III sub-scores of the more affected and less affected sides. IBM SPSS Statistics for Windows, version 25.0 (IBM Corp., Armonk, NY), and R software (version 4.0.3, http://www.r-project.org/) were used to perform all statistical analyses. p < 0.05 was considered statistically significant.
All data are available in the PPMI repository (http://www.ppmi-info.org).
RESULTS
Baseline demographic and clinical characteristics of patients with PD
Fifty-four (13.2%) of 408 patients carried GBA mutations, with the E326K variant being the most common (other mutations are also listed in Table 1), and there was no significant difference in MDS-UPDRS-III scores between the types of GBA variants (Supplementary Table 2). PD patients with GBA mutations were younger than those without mutations (58.91±9.55 vs. 62.10±9.68; p = 0.024). There were no significant differences in sex, disease duration, years of education, the frequency of right side-dominant parkinsonism, and striatal DAT availability between the PD groups according to the presence of GBA mutations. The level of cognitive performance was comparable between the two PD groups except for HVLT-R recognition. There were no significant differences in MDS-UPDRS-III total scores (20.72±8.60 vs. 22.26±10.48, p = 0.307), sub-scores of the more affected side (10.91±4.25 vs. 11.07±3.98, p = 0.786), and sub-scores of the less affected side (3.28±3.39 vs. 4.26±4.85, p = 0.157). Meanwhile, when we compared the estimated MDS-UPDRS-III scores between the groups while adjusting for age, sex, disease duration, and the natural logarithm of SBR in the putamen as covariates, the PD group with GBA mutations had higher estimated MDS-UPDRS-III sub-scores on the less affected side than the PD group without GBA mutations (p = 0.043). The estimated MDS-UPDRS-III total scores and sub-scores of the more affected side did not differ between the groups (p = 0.143 and 0.797, respectively; Tables 12).
Demographic characteristics and striatal DAT availability of patients with PD
Values are expressed as the mean±standard deviation, estimated mean (standard error), or number (percentage). DAT, dopamine transporter; PD, Parkinosn’s disease; GBA(–), PD group without β-glucocerebrosidase (GBA) gene mutations; GBA(+), PD group with GBA gene mutations; MDS-UPDRS-III, Movement Disorders Society Unified Parkinson’s Disease Rating Scale Part III; MoCA, Montreal Cognitive Assessment; Benton’s JLO, Benton’s Judgement of Line Orientation; HVLT-R, Hopkins Verbal Learning Test-Revised. aMDS-UPDRS-III scores were compared between the PD groups using an analysis of covariance while adjusting for age, sex, disease duration, and the natural logarithm of DAT availability in the putamen as covariates. bOne patient for each variant including G115R/G193E, I489L, IVS2 + 1G > A, K(–27)R, N370S/N370S, R44C, R463C, and T369M/R120W.
Effects of glucocerebrosidase gene mutations on the MDS-UPDRS-III scores
MDS-UPDRS-III, Movement Disorders Society Unified Parkinson’s Disease Rating Scale Part III; DAT, dopamine transporter; GBA, β-glucocerebrosidase gene; SE, standard error. aSince the DAT availability in the putamen was not normally distributed, its natural logarithm was used in the general linear model. DAT availability in the more severely affected putamen and less severely affected putamen were used for predicting the MDS-UPDSR-III sub-scores of the more affected side and less affected side, respectively.
Motor reserve according to the presence of GBA mutations
The scatter plots of the natural logarithm of SBR in the putamen (mean, more affected, and less affected sides) and MDS-UPDRS-III scores (total, more affected, and less affected sides) are shown in Fig. 2. For the MDS-UPDRS-III total scores, the prevalence of patients with low motor reserve (motor reserve estimate < 0) or cases above the regression line in the general linear model did not differ between the PD groups according to the presence of GBA mutations (43.2%vs. 46.3%; p = 0.671). Regarding the MDS-UPDRS-III sub-scores of the more affected side and less affected side, the prevalence of patients with low motor reserve also did not differ between the PD groups according to the presence of GBA mutations (46.6%vs. 46.3%, p = 0.966; 38.1%vs. 46.3%, p = 0.253).
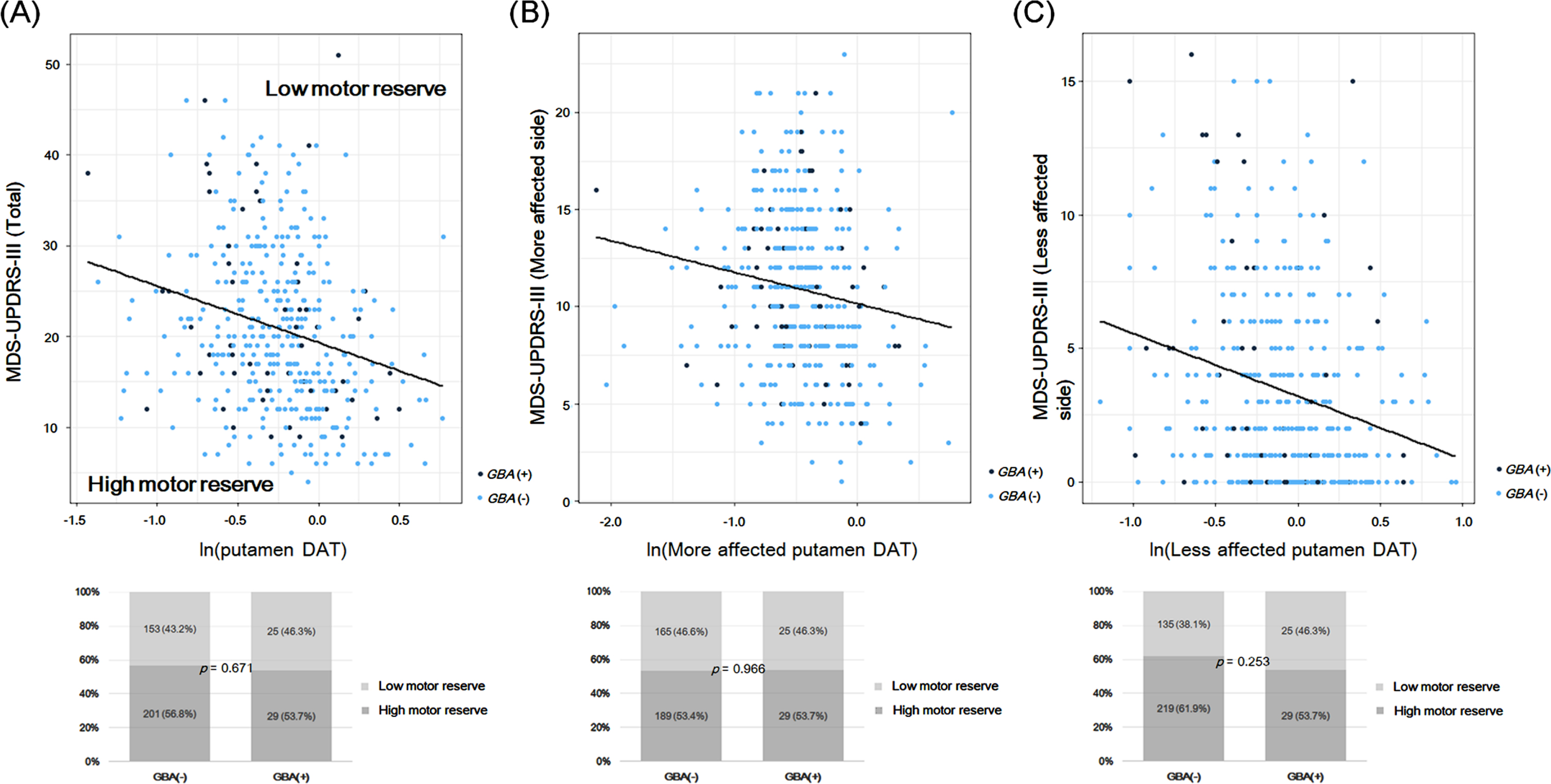
A) Scatter plots of the natural logarithm of SBR in the putamen (mean, more affected side, and less affected side) and MDS-UPDRS-III scores (total, more affected side, and less affected side). The general linear model was used to predict MDS-UPDRS-III scores by using age, sex, disease duration, and the natural logarithm of DAT availability in the putamen. The solid line (black) indicates the regression line of the general linear model. Cases above this line have a higher MDS-UPDRS-III score than the predicted score (i.e., low motor reserve or motor reserve estimate < 0), while cases below this line have a high motor reserve (motor reserve estimate > 0). B) The prevalence of patients with low motor reserve and those with high motor reserve in the PD groups according to GBA mutations. Black circles: PD cases with GBA mutations; Blue circles: PD cases without GBA mutations.
Additionally, we compared the clinical characteristics between the patients with high motor reserve (motor reserve estimate > 0, n = 230) and those with low motor reserve (motor reserve estimate < 0, n = 178; Supplementary Table 3). As expected, the low motor reserve group had greater MDS-UPDRS-III scores than the high motor reserve group after adjusting for striatal DAT binding. There were no significant differences in age, sex, disease duration, and years of education. The high motor reserve group were more likely to be more affected in the right side than the low motor reserve group (62.2%vs. 42.7%, p < 0.001). The high motor reserve group tended to have better performance on several cognitive function tests than the low motor reserve group. There was no difference in the prevalence of GBA mutations between the two groups according to motor reserve estimates (12.6%vs. 14.0%, p = 0.671).
DISCUSSION
The present study investigated whether GBA variants could be a determinant of motor reserve in patients with PD from the PPMI cohort database. The major findings were as follows: 1) Fifty-four (13.2%) patients carried GBA mutations. PD patients with GBA mutations were younger than those without mutations. There were no significant differences in sex, disease duration, years of education, and striatal DAT availability between the PD groups according to the presence of GBA mutations. 2) PD patients with GBA mutations had higher MDS-UPDRS-III scores for the less affected side than those without mutations after adjusting for DAT availability in the contralateral putamen. The MDS-UPDRS-III sub-scores of the more affected side did not differ between the two PD groups. These findings suggested that GBA variants have a detrimental effect on individual capacity to cope with PD-related pathologies (i.e., motor reserve), with different impacts depending on the motor laterality.
Motor symptoms in PD do not develop until the nigrostriatal dopamine is depleted by 60–80%[24], suggesting the presence of compensatory mechanisms in early-stage PD to delay the manifestation of motor deficits. This compensatory capacity appears to vary among patients with PD, and the concept of motor reserve has been proposed to explain the individual differences in the susceptibility to PD-related pathologies [3, 21]. We previously reported several factors that can enhance or reduce the motor reserve in patients with PD [3]. In particular, some modifiable factors including education attainment [4, 25–27], physical activity [5], body mass index [28], and white matter hyperintensity [29], could modulate an individual’s capacity to cope with the pathological changes related to PD. Moreover, although genetic factors associated with motor reserve have not yet been reported, the discovery of genetic factors may help in understanding the underlying pathophysiology and establishing therapeutic strategies to enhance the motor reserve.
The strong association between GBA mutations and PD is well established [30], and glucosylceramidase beta (GCase) deficiency plays biological roles in facilitating PD pathogenesis [12]. Reduction of GCase activity compromises lysosomal protein degradation, subsequently resulting in α-synuclein accumulation [31]. α-Synuclein aggregates inhibit the lysosomal activity of GCase, suggesting the bidirectional effect of α-synuclein and GCase through a positive feedback loop [31]. Mutated GCase protein also leads to endoplasmic reticulum stress and autophagic pertubations [32], as well as mitochondrial dysfunction [33] and microglial activation [34], which may explain why GBA mutations increase the risk of developing PD. Several studies have reported that patients with PD with GBA mutations appear clinically indistinguishable from idiopathic disease at the point of diagnosis [14, 35–37], although they often have a variety of more severe non-motor symptoms [38, 39]. However, in terms of long-term prognosis, patients with PD carrying GBA mutations show a more rapid disease progression of motor impairment and cognitive decline and reduced survival rates compared with those in patients without mutations [13, 40]. These findings suggest that GBA mutations impair the compensatory ability to cope with PD-related pathologies throughout the course of disease. If this is true, GBA mutations could also be a determinant of initial motor reserve (i.e., individual differences in motor deficits despite similar degrees of dopamine neuronal loss) [21] in patients with newly diagnosed PD. Previous studies that could infer the impact of GBA mutations on motor reserve in PD were limited by the lack of DAT scan images at baseline [14, 41] or the fact that some PD patients with GBA mutations were already taking PD medication at recruitment [18, 38]. In particular, Simuni et al. [18] reported that PD patients with GBA mutations at baseline visit had increased SBR values in the putamen contralateral to the more affected side compared with sporadic PD patients at the year 2 visit, which cannot be applicable to the estimation of motor reserve. Meanwhile, our study has the advantage that we estimated the motor reserve of patients with PD who were naïve to PD medication at recruitment while considering DAT availability in the putamen, as in our previous studies [3, 21]. We found that patients with PD carrying GBA mutations had higher MDS-UPDRS-III scores for the less affected side than those without mutations after adjusting for the extent of nigrostriatal degeneration. Genetic variations in GBA might modulate the process of post-synaptic alterations related to motor reserve through pathogenic pathways shared with PD [34]. GBA mutations also might have a detrimental effect on the integrity of striato-cortical pathway [36, 42]. Meanwhile, the level of cognitive performance at baseline except for HVLT-R recognition did not differ between the PD groups according to the presence of GBA mutations, suggesting that cognitive decline reported in other studies at later stages of PD [40, 43] is not evident in recently diagnosed patients with PD with GBA mutations [41].
In terms of motor laterality, the present study showed that patients with PD carrying GBA mutations had lower motor reserve for the less affected side than those with PD without GBA mutations, although the prevalence of patients with low motor reserve according to the arbitrary cut-off for motor reserve estimates did not differ between the PD groups. The motor laterality or asymmetric manifestation of parkinsonism is an important clinical feature of PD [44], with the more-affected body side well correlated with the dopaminergic neuronal loss, which occurs asymmetrically and more severely in the contralateral side of substantia nigra [45]. Several pathomechanisms underlying the asymmetric dopaminergic denervation have been proposed, including an inborn unequal number of substantia nigra neurons between sides, one-sided vulnerability of the blood-brain barrier, and effects of hand dominance [46]. Our findings imply the detrimental effects of GBA mutations on individual capacity to cope with PD-related pathologies, particularly in the less affected side, via the pathogenic microenvironments associated with GBA mutations as discussed above. Meanwhile, no difference in the MDS-UPDRS-III sub-scores on the more affected side might be because nigrostriatal dopamine deficits already reached a critical threshold to manifest parkinsonian motor symptoms and did not allow the influence of GBA mutations on motor deficits for the more affected side in early stages of PD. Interestingly, when we additionally compared the clinical characteristics between the patients with PD with high motor reserve and those with low motor reserve, patients in the high motor reserve group were more affected in the right side than the low motor reserve group, which is in line with our previous work showing that dominant-side patients had greater motor reserve [6]. The high motor reserve group also tended to have higher scores on several cognitive function tests than the low motor reserve group, suggesting a possible link between motor reserve and cognition in patients with PD. These findings suggest that several factors other than GBA mutations also could be determinants of motor reserve in PD.
Our study has some limitations. First, we used SPECT DAT scan imaging data from the PPMI cohort, from which detailed segmentation of the striatum was difficult due to a low spatial resolution to differentiate the subdivisions of the putamen in to the anterior (i.e., associative striatum) and posterior parts (sensorimotor striatum), which have distinct anatomical and functional connections [47, 48]. Second, DAT availability in the putamen may not be the ideal measure for nigrostriatal dopaminergic degeneration [49]. Furthermore, estimation of motor reserve using the residual model could differ between studies, depending on the dataset. Third, although not statistically significant, PD patients with GBA mutations were more likely to have greater MDS-UPDRS-III scores on the right side than those without mutations (59.3%vs. 52.8%), and approximately 90%of patients with PD in the PPMI cohort were right-handed [50]. Therefore, we cannot completely exclude the possibility that the difference in the MDS-UPDRS-III sub-scores on the less affected side might stem from hand dominance, and caution is needed when interpreting the effect of GBA mutations on the motor laterality. Fourth, phenotypic profiles may vary in patients with PD and GBA mutations depending on the variant type (i.e., severe, complex, or mild variants) and GCase activity [38, 51], although it was not feasible to perform subgroup analyses for each variant due to the small sample size in the present study. Fifth, this study was conducted for exploratory purposes based on a relatively small sample size, and multiple comparisons correction for MDS-UPDRS-III sub-scores on the more affected/less affected sides was not performed. Further studies using a larger sample size are needed to draw a firm conclusion.
In summary, the results of the present study demonstrated the detrimental effects of GBA mutations on individual capacity to cope with PD-related pathologies, particularly for the less-affected-sided parkinsonian motor symptoms. These findings suggested that genetic factors could be a determinant of motor reserve in individuals with PD.
Footnotes
ACKNOWLEDGMENTS
Data used in the preparation of this article were obtained from the Parkinson’s Progression Markers Initiative (PPMI) database (![]() ). For up-to-date information on the study, visit www.ppmi-info.org. PPMI (a public-private partnership) is funded by the Michal J. Fox Foundation for Parkinson’s Research and multiple funding partners, including AbbVie, Avid, Biogen, Bristol-Myers Squibb, Covance, GE Healthcare, Genetech, GlaxoSmithKline, Lilly, Lundbeck, Merck, Meso Scale Discovery, Pfizer, Piramal, Roche, Servier, Teva, and UCB. This work was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (grant number: NRF-2021R1I1A1A01059678).
). For up-to-date information on the study, visit www.ppmi-info.org. PPMI (a public-private partnership) is funded by the Michal J. Fox Foundation for Parkinson’s Research and multiple funding partners, including AbbVie, Avid, Biogen, Bristol-Myers Squibb, Covance, GE Healthcare, Genetech, GlaxoSmithKline, Lilly, Lundbeck, Merck, Meso Scale Discovery, Pfizer, Piramal, Roche, Servier, Teva, and UCB. This work was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (grant number: NRF-2021R1I1A1A01059678).
CONFLICT OF INTEREST
The authors have no conflict of interest to report.