
Abstract
Recent data support an involvement of defects in homeostasis of phosphoinositides (PIPs) in the pathophysiology of Parkinson’s disease (PD). Genetic mutations have been identified in genes encoding for PIP-regulating and PIP-interacting proteins, that are associated with familial and sporadic PD. Many of these proteins are implicated in vesicular membrane trafficking, mechanisms that were recently highlighted for their close associations with PD. PIPs are phosphorylated forms of the membrane phospholipid, phosphatidylinositol. Their composition in the vesicle’s membrane of origin, as well as membrane of destination, controls vesicular membrane trafficking. We review the converging evidence that points to the involvement of PIPs in PD. The review describes PD- and PIP-associated proteins implicated in clathrin-mediated endocytosis and autophagy, and highlights the involvement of α-synuclein in these mechanisms.
BACKGROUND
Parkinson’s disease (PD) is a heterogeneous neurodegeneration. Disease heterogeneity is reflected both at the clinical and molecular levels. PD is generally considered a sporadic disease of unknown etiology. Nevertheless, in the past two decades, it has become apparent that many cases (about 5–10%) are due to familial, inherited forms of the disease [1]. A growing list of genes and loci has been genetically implicated in the pathogenesis of PD. Classifying these genes, according to their known functions, provides valuable clues about the cellular mechanisms involved in PD. A mechanistic role in vesicular membrane trafficking was recently highlighted for many of the PD-associated proteins (recently reviewed in [2]). The available genetic data also support a role for phosphoinositides (PIP) in the pathophysiology of PD. Central PD-associated genes encode for proteins that regulate PIP homeostasis at cell membranes. Other PD-associated genes encode for proteins that directly interact with PIPs or other PIP-binding, or PIP-metabolizing proteins (Table 1). This review focuses on PD- and PIP-associating proteins that play a role in vesicular membrane trafficking, in particular, clathrin-mediated endocytosis and macroautophagy.
PD-associated proteins and their mode of PIP-associations
PIPs are phosphorylated derivatives of the acidic membrane phospholipid, phosphatidylinositol (PI). The seven PIP molecules differ in the position and number of phosphorylated hydroxyls on the inositol ring of this phospholipid. All PIP members are master regulators of cellular signaling pathways [3]. They are distributed between cell membranes in a defined and characteristic manner that is maintained locally by PIP-kinases and PIP-phosphatases, which generally present specificity toward the phosphate(s) position on the inositol ring. The composition of PIPs on cell membranes is also dependent on cell activities, vesicular trafficking, and non-vesicular lipid transport [4]. Accordingly, phosphatidylinositol 3 phosphate (PI3P) is enriched at early endosomes; phosphatidylinositol 4 phosphate (PI4P) at Golgi membranes, the plasma membrane and recycling endosomes; phosphatidylinositol 3,5 bisphosphate (PI3,5P2) at late endocytic compartments; phosphatidylinositol 4,5 bisphosphate (PI4,5P2) and phosphatidylinositol 3,4,5 triphosphate (PI3,4,5P3) are enriched at the plasma membrane [3–6].
The defined and characteristic PIP composition on subcellular membranes provides a lipid signature that directs proteins to specific organelles and facilitates cell activity. Proteins may interact with PIPs through direct binding, mediated by high-affinity PIP-binding domains or charge attractions, formed between the acidic PIPs and positively charged protein domains. PIP-binding proteins may recruit other proteins to the membrane through protein-protein interactions. In addition to directing proteins to specific organelles, the interaction with PIPs may allosterically activate proteins [3].
PI, the precursor of all PIPs, is synthesized at the endoplasmic reticulum. It is then delivered to cell membranes by non-vesicular lipid transport at membrane contact sites (MCSs) [6, 7] and by vesicular trafficking. PI is an abundant membrane phospholipid, representing ∼10–20 mol % among mammalian membranes. However, the total levels of PIPs appear to be maintained at very low levels, estimated at 2–5% of PI [4], suggesting that the regulation of PIP homeostasis may involve additional factors that are yet to be identified.
Table 1 lists PD-associated proteins that are linked with PIPs through one of the following categories: 1) PD-associated genes involved in homeostasis of PIPs, 2) PD-associated genes that directly interact with PIPs, or 3) PD-associated genes that interact with other PIP-binding proteins. The genetic evidence linking these genes with PD includes identified mutations with Mendelian PD inheritance or contributing risk factors identified in genome-wide association studies (GWAS). The genes listed in Table 1 are involved in clathrin-mediated endocytosis and/or macroautophagy, mechanisms consisting of vesicular membrane trafficking.
PIPs AND VESICULAR MEMBRANE TRAFFICKING
Cell membranes regulate vital mechanisms of cell biology and adequate responses to external and internal stimuli. The membranes undergo continuous adjustments of their contents, including phospholipids and fatty-acyl side chains, to enable structural and functional membrane plasticity [8]. The dynamic nature of cell membranes underlies vesicular membrane trafficking and facilitates cargo delivery, including proteins and other molecules wrapped up in double-membrane vesicular structures, between cell compartments or between the cell and its environment. Vesicular membrane trafficking is mediated through vesicles budding from a donor membrane and fusing with a destination membrane. It involves the recruitment of specific membrane-interacting proteins to support and stabilize defined stages in the process. Major vesicular trafficking routes include: 1) the secretary ER to Golgi pathway that delivers folded and post translationally modified proteins, as well as additional cargo molecules, through COPII and COPI-coated vesicles; 2) internalization of molecules from cell exterior into the cell interior by clathrin-coated or other membrane vesicles and then sorting by endosomes; 3) the internalized cargo can be delivered to the lysosomes for degradation or recycled back to the plasma membrane; 4) the retromer complex rescues proteins from lysosomal degradation by diverting them to the retrograde endosome-to-Golgi trafficking pathway or back to the plasma membrane through a recycling pathway [9]; and 5) macroautophagy engulfs a portion of the cellular contents in autophagosome for delivery to the lysosome for degradation [10]. Specific adaptations of vesicular membrane trafficking mechanisms enable specialized cell activities. For example, neuronal cells communicate with their environment through exocytosis of synaptic vesicles (SVs) and release of neurotransmitters. Exocytosis of SVs is coupled with mechanisms of endocytosis, including endocytosis mediated by the formation of clathrin-coated vesicles [11, 12].
PIPs play critical roles in vesicular membrane trafficking [3, 13]. The defined and characteristic composition of PIP on cell membranes enables the formation of a gradient of PIP-concentrations, along which the levels of specific PIPs in the origin membrane of the vesicle are altered to adjust with vesicle development and the composition of PIPs on the target membrane. The process of formation and development of membrane vesicles is accompanied by a coordinated recruitment of PIP-kinases and/or PIP-phosphatase to enable the required transitions in the composition of PIPs and to facilitate the fusion with the target membrane [5].
CLATHRIN-MEDIATED ENDOCYTOSIS (CME)
CME is the major endocytic pathway in mammalian cells (for recent reviews [14, 15]). This mechanism regulates the partitioning of transmembrane transporters and receptor proteins between the surface of the cell and the interior. In this way, CME regulates the availability of these proteins to receiving and internalizing signals and molecules from the external environment into the cell (Fig. 1). CME occurs through clathrin-coated pits (CCPs). A curve-forming membrane-structure coated with a clathrin lattice, which comprises three clathrin heavy chains (CHC), with tightly associated clathrin light chains (CLC) and the adaptor protein-2 (AP2), a heterotetramer consisting of α, β2, μ2, and σ2 subunits. CME can be dissected into four stages: (a) initiation, (b) stabilization, (c) maturation, and (d) membrane scission. The released clathrin-coated vesicle (CCVs) is then uncoated and delivers its cargo to early endosomes. Specific endocytic accessory proteins function to facilitate each stage and the transition between the stages. Importantly, this orchestrated transition between stages is critically regulated by PIPs [14–16]. Sequential recruitment of specific PIP-kinases and PIP-phosphatases generates the spatial and temporal combination of PIPs required to support the process. At initiation on the plasma membrane, PI4,5P2 is generated from PI4P by phosphatidylinositol 4-phosphate 5-kinase 1 (PIPKI). It acts to recruit the endocytic clathrin adaptors and their accessory factors to initiate the formation of a CCP [17, 18]. The maturation of the vesicle is accompanied by a gradual decline of PI4,5P2 and generation of PI3,4P2 in a two steps process where PI4,5P2 is first converted to PI4P by removal of the phosphate from the 5th hydroxyl position on the myo-inositol ring, a process mediated by synaptojanin1 (SJ1) [19] and SHIP2, inositol-5-phosphatase [20]; PI4P is then phosphorylated at the 3rd position of the inositol ring to generate PI3,4P2 by the class II phosphatidylinositol 3-kinase C2α (PIK3C2α) [21]. PI3,4P2 facilitates the recruitment of Bin/Amphiphysin/Rvs (BAR) domain-containing proteins, including sorting nexin (SNX)9/18 and vesicle scission by dynamin [5]. Vesicle uncoating occurs post scission in a process that requires dephosphorylation of PI4,5P2 to PI4P and synthesis of PI3P on the vesicle [22] to facilitate fusion with early endosomes [13]. A certain degree of inconsistency, attributed to differences in tracing molecules or other technologies, persists [5, 15]. The importance of dephosphorylating PI4,5P2 to PI4P for vesicle maturation was initially identified in CME of SVs in neuronal synapses and was shown to depend on the PI4,5P2-phosphatase, SJ1 [23]. Neurons of mice, in which the brain-specific SJ1 isoform was knocked out, accumulated coated SVs as well as PI4,5P2 [24]. In non-neuronal cells, the 5-phosphatase OCRL is implicated in PI4,5P2 dephosphorylation during endocytosis [25, 26]. OCRL is suggested to function in close cooperation with the 4-phosphatase, Sac2/INPP5F [27]. A cooperative function of OCRL/Sac2/INPP5F presents dual 5- and 4-phosphatase activities similar to the neuronal SJ1 [27].
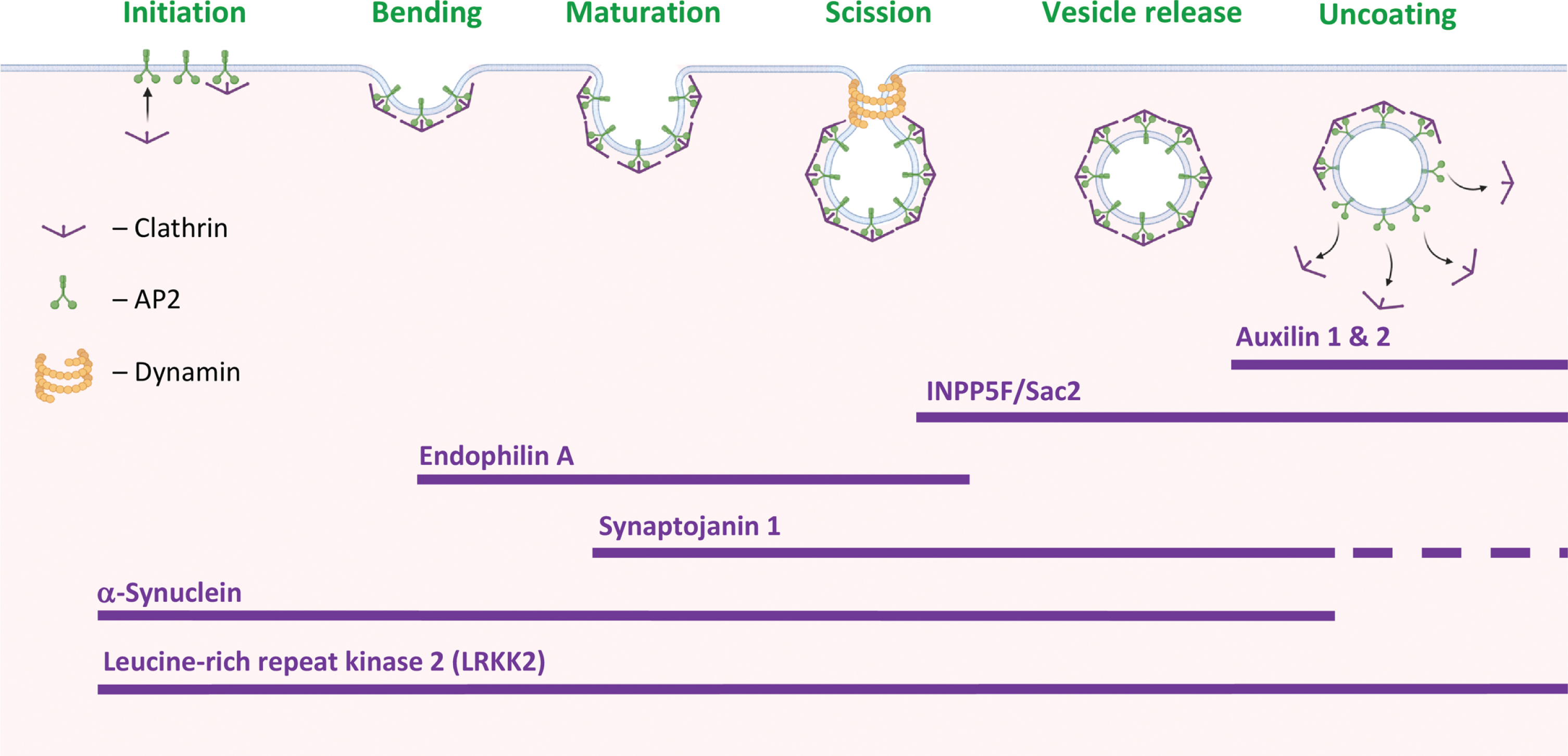
PD-proteins in clathrin-mediated endocytosis. Stages in clathrin-mediated endocytosis (CME) and the suggested involvement of PD-associated proteins at specific stages of the process. Created with BioRender.com.
An involvement in CME has been attributed to several PD-associated proteins (Fig. 1). From the initiation and stabilization of the CCP to vesicle scission. That is, α-Synuclein (α-Syn) and Leucine-rich repeat kinase 2 (LRRK2) are involved throughout the process; Endophilin A, in CCP membrane bending and invagination; SJ1, in CCP maturation; INPP5F/Sac2 and SJ1, in vesicle scission and release; and auxilin 1 & 2, in vesicle uncoating.
AUTOPHAGY
Autophagy regulates the recycling of cellular components by degrading dysfunctional or damaged proteins and organelles. There are three forms of autophagy: macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA) [10]. In macroautophagy, a membrane-based structure called phagophore, engulfs a portion of the cytoplasm and gradually develops into a mature autophagosome vesicle. The autophagosome membrane will then fuse with membranes of endosomes or lysosomes [28]. In microautophagy, an endosome or lysosome directly invaginates and engulfs its cargo. The invaginated membrane pinches off into the lumen of its originating organelle as a microautophagic body [29]. In CMA, the cargo is selectively recognized by a chaperone protein and then directly internalized into the lysosome for its degradation [30]. Microautophagy and CMA, depend on cognate protein of 70 kDa (Hsc70) for target degradation. In addition, CMA requires the lysosome-associated membrane protein 2 isoform A (Lamp2A) as protein receptor [30, 31]. Thus, macroautophagy operates through vesicular trafficking, however, microautophagy and CMA proceed with a direct engulfment of cargo into the endolysosomal compartment [32]. Evidence for increased abundance of autophagosomes in brains with PD [33, 34] and the abundance of lysosomes in the Lewy-bodies [35] directly connect the autophagy-lysosomal pathway with the disease. Although it is not clear yet whether the data indicate increased clearance of damaged proteins and organelles by autophagy or defective autophagic mechanisms.
PIPs are critically involved in virtually every step in the autophagy process (for a recent review see [36, 37]). It is now generally agreed that PI3P plays a pivotal role in the initiation of autophagy; PI4P and PI4,5P2 are required for the gradual growth of the phagophore and generation of a double- membraned autophagosome; and PI3,5P2 is important for the fusion of the autophagosome with the lysosome, to form an autolysosome. To achieve the fine dynamic balance in membrane PIP composition along the process, specific PIP-interacting proteins or PIP-modifying enzymes are sequentially recruited throughout the process [36, 38]. In mammalian cells, the key signal to suppress or initiate autophagy predominantly relies on the mammalian target of rapamycin complex 1 (mTORC1), which is regulated by PM levels of PI4,5P2. Conversion of PI4,5P2 to PI3,4,5P3 by PI3 kinase is leading to activation of mTORC1 and inhibition of autophagy. The reverse effect of PTEN 3-phosphatase, to dephosphorylate PIP3 and generate PI4,5P2, initiates the autophagy process.
PIPs also regulate autophagic lysosome reformation, an alternative pathway for lysosome generation during autophagy, in which a localized budding of autolysosome membranes forms tubules that undergo scission to generate new lysosomes [39, 40]. This process is facilitated by PI4P and PI4,5P2 that recruit protein effectors to the reformed membranes. The inositol polyphosphate 5-phosphatase, INPP5K, is required for this process [41]. Mutations in INPP5K are associated with marked lysosome depletion and inhibition of autophagy. Resulting from the reduced conversion of PI4,5P2 to PI4P on autolysosomes, and impaired autophagic lysosome reformation [42, 43]. In relevance to this review, INPP5K was identified as a contributing risk factor for PD [44, 45].
The critical relevance of autophagy in familial and sporadic PD was recently reinforced by the discovery of mutations in specific PD-associated genes and contributing risk factors encoding for proteins that are part of the autophagy-lysosomal and mitophagy pathways (recently reviewed [46–48] and Table 1). In addition, histopathological evidence has pointed at the accumulation and defective clearance of autophagosomes in PD brains [48]. Protein markers of autophagosomes and lysosomes were commonly detected within Lewy-pathology of PD brains, as well as the related synucleinopathies, multiple system atrophy and dementia with Lewy-bodies [35, 48]. Together, autophagy is implicated in PD at the genetics and histopathology of the disease.
PIPs, AUTOPHAGY AND CME IN PD
Autophagy and endocytosis are two distinct vesicular membrane trafficking mechanisms that intersect at several stages throughout vesicle formation, transport and fusion. These mechanisms share components of the molecular machinery and a common endpoint at the lysosome (reviewed in [31, 49]). Growing evidence suggests that autophagy is dependent on endocytosis and vice versa [31]. Many of the PD- and PIP-associated genes are involved either in CME or autophagy, of which several genes including, α-Syn, SJ1, endophilin A, LRRK2 and members of the Rabs family, play a role in both mechanisms (Fig. 2).
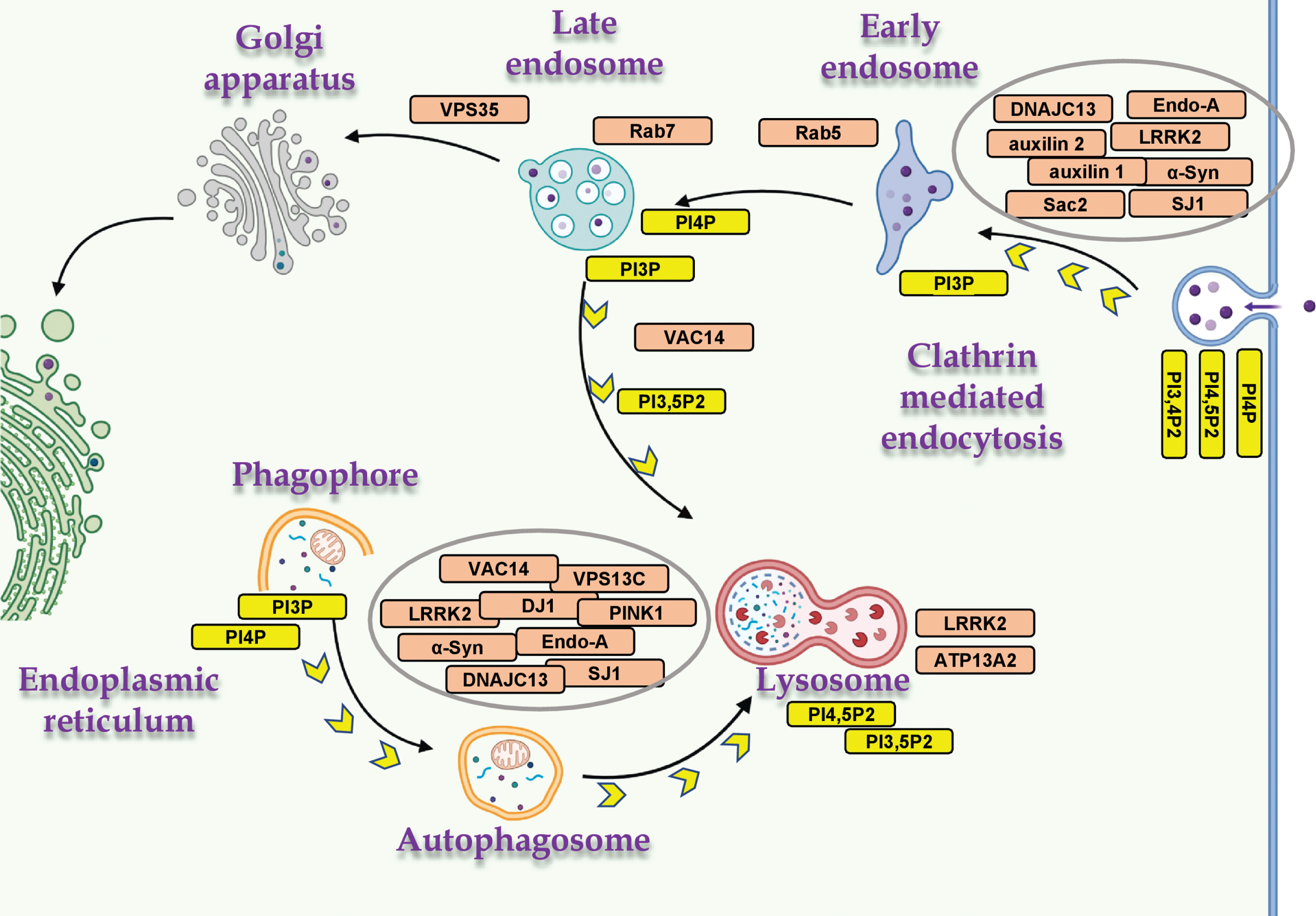
PD- and PIP-associated proteins in vesicular trafficking. The distribution of PD- and PIP-associated proteins in cellular mechanisms of vesicular membrane trafficking, and the relevant PIPs. Proteins involved in both mechanisms, macroautophagy and CME (α-Syn, LRRK2, SJ1 and endophilin A); mitophagy (PINK1 and DJ1); and endo-lysosomal proteins (ATP13A2, VAC14, VPS35, Rab5 and Rab7). Created with BioRender.com.
SJ1
SJ1 and PD
SJ1 harbors two PIP-phosphatases. A 5-phosphatase that dephosphorylates PI4,5P2 and also PI3,4,5P3 [23, 50], and a Sac1 PI 4-phosphatase that dephosphorylates PI4P to PI and can also function as a PI-3 phosphatase [23, 51]. Mutations in the Sac domain (R258Q, R459P) or the 5-phosphatase domain (Y832C, R839C) of SJ1 have been associated with early-onset Parkinsonism and typical PD, respectively [52–55], and defined SJ1 as PARK20 [54, 56–60]. Data obtained from genome-wide expression studies reported down-regulation of SJ1 transcript in brains affected with sporadic PD [53]. Knock-in mice, carrying the corresponding human R258Q mutation in the Sac1 domain of the mouse SJ1 [52], showed motor defects and epilepsy. In relevance to PD, the mice demonstrated structural alterations in dopaminergic nerve terminals in their dorsal striatum [52]. In addition, a reduced rate of SVs endocytosis accompanied by abnormal accumulations of clathrin-coated intermediates, was reported. Based on these findings, a role for the Sac domain of SJ1 in vesicle uncoating was suggested. The R258Q mutation in SJ1 was shown to abolish the phosphatase activity of the Sac domain without affecting the 5-phosphatase activity [54]. This finding supports an independent activity for each of SJ1 phosphatases in CME. Together, the PD-associated mutations in SJ1 link CME and disease mechanisms [52]. An additional mouse model, carrying a heterozygous deletion of SJ1 (SJ1+/-) displayed an age-dependent motor dysfunction, impaired SV endocytosis and degeneration of dopaminergic terminals [53].
SJ1, PIP and CME
A well-defined function of SJ1 in the synapse, is to couple SV endocytosis with dephosphorylation of PI4,5P2, by sequentially removing the phosphates from the 5th and 4th positions of the inositol ring via its two phosphatase domains [13, 61]. Two splice variants for SJ1 are known, a 145- and a 170-kDa isoforms [23]. Although both isoforms are ubiquitously expressed, SJ1-145 is present at very high concentrations in presynaptic nerve terminals of the adult brain [62]. The SJ1-170 splice variant contains binding sites for clathrin and the clathrin-adaptor AP2 and is present throughout the formation and maturation of CCPs [19]. Whereas, the SJ1-145 variant is involved in the dephosphorylation of PI4,5P2 as part of vesicle scission and uncoating [19].
Recruitment of SJ1 to endocytic sites is mediated by the PD-associated endocytic protein, endophilin [63, 64]. Genetic deletion of either SJ1 or endophilin in mouse neurons leads to defects in synaptic transmission [24, 66], represented by a lower abundance of SVs in nerve terminals; delayed SV endocytic recycling and accumulation of coated vesicles that have not gone through the uncoating step of CME.
SJ1 and autophagy
While SJ1 has long been known for its involvement in SV cycling, recent data support a role for SJ1 in autophagy in the presynaptic terminals [67, 68]. A study in a knock-in Drosophila model, expressing the equivalent to the human R258Q mutation in the endogenous SJ1 Sac domain (R228Q), reported interference with the 3-phosphatase activity [67] and accumulation of synaptic autophagosomes. The accumulated autophagosomes were positive to WIPI2/Atg18 protein, which is a PI3P/PI3,5P2-binding protein, suggesting abnormal PIP homeostasis at nerve terminals. In addition, abnormal maturation of autophagosomes was detected in patient-derived induced neurons, carrying the human SJ1 R258Q mutation [67]. Impaired autophagy was also reported in mice carrying a heterozygous deletion of SJ1 (SJ1+/-) [53]. These results suggest that the PIP-phosphatase activities of SJ1 couple autophagy with vesicle cycling and synaptic activity.
Of relevance, a second PIP-phosphatase, Sac2/INPP5F, containing a Sac domain that functions primarily as a PI4P phosphatase [27, 69], was located within a PD risk locus identified by GWAS [70]. Sac2/INPP5F has been localized to vesicles formed by CME, as well as other endocytic membranes, including macropinosomes, and Rab5 endosomes [27, 69]. A synergistic role for Sac2/INPP5F and SJ1 in regulating PI4P pool size, whose dysfunction results in PD, was suggested [27, 71].
α-Syn
α-Syn and PD
α-Syn is strongly implicated in the genetics of PD. GWAS data showed that the SNCA gene, encoding for α-Syn protein, makes the largest contribution to the genetic risk associated with idiopathic PD [72–74]. Genetic variability within the SNCA promoter that results in higher expression levels of α-Syn is associated with idiopathic PD [75]; Higher α-Syn expression levels, resulting from gene duplications or triplications of the chromosomal loci encoding SNCA, are a dominant-inherited cause of PD [76–78]. Moreover, six autosomal dominant mutations in SNCA have been described, namely, A53T [79] A30P [80], E46K [81], H50Q [82, 83], G51D [84, 85] and A53E [86, 87].
α-Syn is also implicated in the histopathology of PD. α-Syn is a major constituent of Lewy-pathology [88], the hallmark pathology of PD, including Lewy-bodies and Lewy-neurites [89]. It is currently debated whether in the Lewy-bodies α-Syn occurs primarily in fibrillated or non-fibrillated forms [35, 90]. A debate with a significant impact on the way we understand the pathogenesis of α-Syn in PD and the related synucleinopathies [91].
α-Syn, membrane phospholipids and PIP
Early studies have shown that α-Syn binds membrane lipids [92] with a preference for negatively charged phospholipids [92–94]. A lipid-binding domain in α-Syn protein, consisting of a stretch of ∼100 aa residues at its N-terminus [94], harbors seven imperfect repeats of a KTKEGV motif [95]. Positively charged Lysine residues within the repeat motif were shown to be essential for lipid binding, mediated by charge attraction with the negatively charged phospholipid head groups [96, 97]. Membrane binding enhances structure acquisition for this intrinsically disordered protein [92, 99]. The membrane binding domain forms an extended curved α-helical structure [100] that promotes binding to curved membranes, akin vesicular structures [101]. A hydrophobic core region within the membrane binding domain is suggested to penetrate the phospholipid bilayer beyond the plain of the phospholipids head-group, where it interacts with their fatty acyl chains [102–104]. It has been suggested that the membrane binding domain in α-Syn senses membrane curvature; facilitates the formation or stabilizes a curvature on membrane structures [105]; or forms a tether between vesicles [101]. The acidic C-terminal region in α-Syn has been suggested to support and sterically stabilize curved membrane structures [106]. In accord with these reports, a role for α-Syn in the maintenance [107, 108] and dynamics of SV pools has been reported [109, 110].
Additional sets of data support an involvement of α-Syn in regulating the content of lipids on cell membranes. Previous studies have shown that α-Syn binds fatty acids; enhances their uptake into cell lines and mouse brains; and incorporation into phospholipids [111, 112]. A preference for uptake and metabolism of polyunsaturated fatty acids (PUFA) was suggested [111, 112]. A recent study reported increases in levels and metabolism of Oleic acid, a mono-unsaturated fatty-acid, upon overexpression of α-Syn in yeast cells, mouse neurons and human induced pluripotent stem cell (iPSC)- neurons [113]. Lipidomic data in yeast cells further showed alterations in the profiles of phospholipids, triglycerides and cholesterol with α-Syn expression [113]. α-Syn effects on membranes’ phospholipid content were also demonstrated in myelin membranes purified from α-Syn tg mouse brains. Higher levels of phospholipids, detected by 31P NMR spectra, were reported in young and healthy α-Syn tg mice [114]. In accord with the increases in the content of lipids, accumulation of lipid droplets was detected with α-Syn expression [113, 116]. Importantly, the alterations in lipid content observed in the disease models were shown to associate with α-Syn toxicity [113, 117–121]. Together, the data support a general role for α-Syn in lipid homeostasis and metabolism.
In relevance to disease mechanisms, alterations in lipid profiles were reported in brains affected with PD and multiple system atrophy at autopsy (reviewed in [97, 123]. Importantly, genetic studies pointed at several genes that are involved in lipid metabolism and implicated in the genetics of PD, including biosynthesis and turnover of phospholipids, glycolipids, sphingolipids and fatty acids [97].
Based on two main observations relevant to α-Syn associations with lipids, its preference for binding acidic phospholipids and its effects to increase the content of membrane phospholipids, it was hypothesized that α-Syn associates with PIPs. Indeed, it was found that α-Syn specifically interacts with PIPs [124–128]. The data further suggested that α-Syn expression increased the steady-state levels of PI4P, PI3,4P2 and PI4,5P2 [128]. Expression of the PD-associated mutations in α-Syn, A53T and A30P, further increased membrane levels of these PIPs [128]. A mutant form of α-Syn, which denies lipid binding, due to charge replacement in the KTKEGV repeat motif, was ineffective in this respect [96, 128], suggesting that this motif is critical for α-Syn regulation of PIPs. Of interest, in relevance to α-Syn effect to enhance uptake and incorporation of PUFAs [111, 112], PIPs are normally enriched with polyunsaturated fatty acyl chains [4].
The mechanism through which α-Syn acts to regulate the overall content of membrane lipid is currently unknown. Such a mechanism may potentially involve alterations in the expression levels of genes that are master regulators of lipid synthesis and homeostasis [129, 130]. A potential explanation may involve α-Syn translocation to the nucleus and its involvement in transcription activation mediated by nuclear receptors [95, 131], including regulators of lipid homeostasis [132, 133]. In relevance to this review article, although no mechanistic explanation currently supports α-Syn involvement in the regulation of PIPs, the available data place α-Syn among PD- and PIP-associating protein (Table 1).
α-Syn, CME and SV cycling
A role for α-Syn in CME was first suggested based on the determination of transferrin’s internalization, a CME prototype, in cultured dopaminergic cells [134]. α-Syn-mediated enrichment of cell membranes with PUFAs enhanced membrane fluidity [112] and further enhanced endocytosis of transferrin [134]. Accordingly, enrichment of membrane phospholipids with PUFA facilitated membrane invagination and scission of membrane vesicles mediated by endophilin and dynamin proteins [135]. The reported enrichment of α-Syn expression at presynaptic terminals [95, 136] and the knowledge that CME is a major route for SVs endocytosis, supported a potential role for α-Syn in SVs cycling. Indeed, loading the synapses with the lipophilic FM1-43 dye following evoked synaptic activity indicated lower uptake in cultured hippocampal neurons from α-Syn-/- than in wild type mouse neurons [134]. Restoration of α-Syn expression in primary cortical neurons from α-Syn-/- mouse brain enhanced endocytosis determined by the pH-sensitive fluorescence of Synaptophysin-pHluorin (SypHy) chimeric indicator [128]. It was further suggested based on α, β and γ-Syn knock-out in mice that all three synucleins are involved in clathrin-mediated SV recycling at presynaptic nerve terminals [137].
Growing evidence now supports a role for α-Syn to facilitate and increase the rate of endocytosis [128, 137–139]. α-Syn colocalizes with clathrin [134, 137]. Its colocalization with phosphorylated AP2, PI4,5P2 and PI3,4P2 [128] supports its recruitment to CME already at the initiation stage. Data obtained in synapses of dynamin 1, 3 KO neurons, in which CME is arrested at the scission step and synapses accumulate clathrin pits [140, 141], show that α-Syn is colocalizing with clathrin in the arrested clathrin pits [137]. Moreover, α-Syn was shown to directly interact with Hsc70 and excess of α-Syn at the lamprey reticulospinal synapse leads to sequestration of Hsc70 and impairment of CCV uncoating at the synapse [142]. Together, these findings support an association of α-Syn with CCPs from vesicle initiation, throughout their growth and maturation, to scission and uncoating. However, other studies reported that excess of α-Syn in the synapse interfered with endocytosis [124, 143]. Moreover, loading synapses of the lamprey neurons with α-Syn resulted in inhibition of SV endocytosis during intense electrical stimulation [144]; and neurons overexpressing α-Syn were shown to internalize lower amounts of styryl dye indicators for SV recycling, indicating a reduction in endocytosis [109, 145].
Alongside the reports describing a role for α-Syn in CME and SV endocytosis, a large body of evidence also supports a role for α-Syn in SV exocytosis (recently reviewed in [146, 147]). α-Syn was shown to play roles in the assembly of the SNARE complex [148–151]; vesicle docking and fusion [152, 153]; transmitter release [110, 155]; and regulation of fusion pore dilation [156]. To summarize a large body of evidence that links α-Syn with vesicular membrane trafficking, it is currently difficult to conclude whether α-Syn enhances or inhibits the process, and whether it is endocytosis or exocytosis [146, 147].
A recent study has shown that the actual composition of PIPs in the plasma membrane determines α-Syn’s activity in the endocytosis of transferrin. A rapid recruitment of PIP 5-phosphatase to the plasma membrane [157], acutely depleted PI4,5P2 content and in accord, inhibited endocytosis of transferrin. The data further supported a role for membrane PIPs in SV endocytosis mediated by α-Syn [128]. Importantly, in addition to its roles in SVs endocytosis, PI4,5P2 is critically involved in SVs exocytosis. Including, priming and fusion steps of Ca2 + -triggered vesicle release [158]; recruitment and activation of specific protein regulators of SNARE complex assembly and function [158, 159]; and involvement in dilation of the fusion pore [158, 159]. PI4,5P2 roles in exocytosis may also involve PI4,5P2-binding proteins such as CAPS, Munc13 and synaptotagmin [160–163] or its effects on F-actin polymerization [164].
Considering the findings showing that α-Syn involvement in SVs cycling is mediated through its effects to enrich cell membranes with PIPs, it is reasonable to hypothesize that similar to its effects in endocytosis, α-Syn may also act in exocytosis by enriching the presynaptic membranes with PIPs.
It is thus important to better understand the involvement of α-Syn in the regulation of synaptic PI4,5P2 levels. Whether there are single or multiple pools of PI4,5P2 at the synapse and whether regulatory checkpoints may act to maintain a balance in PIP availability for the endocytic and exocytic arms of SV cycling.
α-Syn and autophagy
The associations of α-Syn with autophagy are complex. Toxic α-Syn forms, including misfolded or aggregated α-Syn, are cleared by CMA [165, 166] and macroautophagy [167]. In relevance to the clearance of α-Syn, data suggest the occurrence of a cellular cross-talk between macroautophagy and CMA. A compromise in CMA results in activation of macroautophagy and vice versa [30]. Additional data support an involvement of α-Syn in macroautophagy, yet with a certain degree of controversy. That is, over expression of α-Syn was shown to enhance macroautophagy [165, 169]. However, other studies have reported evidence showing an inhibitory role for α-Syn in autophagosome formation and macroautophagy [170, 171].
In summary, the controversies over α-Syn effects in autophagy and CME, may relate to one another. The shared components between the two pathways, including the dependence on vesicular membrane trafficking and the critical roles of PIPs throughout the processes, bring up a question related to the mechanism of action of α-Syn in autophagy. Whether similar to its role in CME, α-Syn may act in autophagy to alter the steady state levels of specific membrane PIPs?
LRRK2
LRRK2 and PD
LRRK2 is a large multidomain protein harboring a kinase, GTPase and protein-binding domains [172]. Missense mutations in LRRK2 cause late-onset autosomal dominant PD which is clinically indistinguishable from sporadic PD [173, 174]. LRRK2 mutations are the most common genetic cause of familial PD [173, 175] and GWAS studies identified LRRK2 as a risk factor for idiopathic PD [70]. Pathologically, LRRK2 mutation carriers experience progressive neurodegeneration of the nigrostriatal pathway and often develop α-Syn-positive Lewy-bodies. In addition, a subset of patients with LRRK2 mutations presents inclusion bodies that are negative for α-Syn yet, positive for tau and TDP-43 [173, 176–178]. Suggesting that the pathophysiology of LRRK2 in PD may occur independently of classical Lewy-pathology. Through its kinase activity, LRRK2 interacts with many PD-associated proteins. In addition, Rab29 [179–181] and VPS35 [181, 182] proteins associated with PD, are upstream regulators of LRRK2. Due to its central roles in the genetics and pathophysiology of PD, LRRK2 is a subject of intensive research. The readers are referred to other recent reviews that provide a comprehensive description of the current knowledge (for example [172, 183]).
LRRK2 and PIP
While no evidence is currently known to directly link LRRK2 and PIPs, ample evidence shows that LRRK2 interacts with and phosphorylates PD-associated, PIP-interacting proteins. These include the following proteins that are involved in vesicular membrane trafficking: ATP13A2; Auxilin 1, and 2; Dynamin; endophilin-A1; SJ1; VPS35; and specific Rab proteins (see Table 1 and below).
LRRK2 and CME
LRRK2 plays a role in CME and SV endocytosis. Silencing LRRK2 expression in flies or mice resulted in accumulation of endocytic intermediates; an abnormal vesicle morphology; and a reduced number of synaptic vesicles [184, 185]. Notably, the defects in LRRK2 activity in endocytosis were attributed to its kinase activity [185]. The most common LRRK2 G2019S PD-mutation was shown to impair SV endocytosis in mouse ventral midbrain neurons, including in dopaminergic neurons [186]. It was further shown that the impairments in endocytosis, resulting from LRRK2 G2019S transgenic expression, could be rescued by inhibiting LRRK2 kinase activity [186, 187]. Reductions in SV densities were also reported for iPSC-derived dopaminergic neurons expressing the LRRK2 R1441C mutation [188]. Moreover, the vesicles appeared to lack distinct surrounding membranes, consistent with membrane-less clathrin cages observed in models involving defective clathrin uncoating [188].
LRRK2 was shown to interact physically and functionally with critical components or facilitators of CME and SV endocytosis, including auxilin [188], Dynamin 1, 2, and 3 [189], SJ1 [186, 190] and endophilin-A1 [184, 185]. LRRK2 also interacts with clathrin [191] and the clathrin adaptor AP2 complex, regulating the recruitment of AP2 [187] and phosphorylation of AP2M1 at threonine 156 [192]. Finally, data show evidence for indirect associations between α-Syn and LRRK2 [177]. Together, the reported data suggest that, through its phosphorylation and other associations with proteins involved in CME, LRRK2 is a master regulator of CME, recruited to the process at the early stage of initiation and supports the process throughout, to vesicle uncoating (Fig. 1).
LRRK2 and autophagy
An involvement for LRRK2 in autophagy is now well documented (reviewed in [172, 193]). However, inconsistencies in autophagy outcome, whether increasing or decreasing the autophagic flux, persists [172, 183]. The inconsistencies are explained by differences in experimental models and deny a conclusion about the mechanism of action of LRRK2 in autophagy.
Similar to α-Syn, LRRK2 is a substrate for CMA [165, 194]. The PD-associated G2019S mutation in LRRK2 and high levels of wild type LRRK2 were shown to inhibit CMA [194–197]. CMA inhibition by LRRK2 interfered with α-Syn clearance [194, 198], resulting in intracellular accumulation of α-Syn. The G2019S mutation increases LRRK2 kinase activity [199–201]. Interestingly, inhibitors for LRRK2 kinase activity were shown to reduce the accumulation of α-Syn in human neuronal cell lines carrying the G2019S mutation [202]. Supporting a pathogenic role for LRRK2 that involves α-Syn toxicity in PD.
LRRK2 was found to form a complex with Rab7L1 and auxilin 2 proteins, encoded by genes identified as risk genes for sporadic PD, and BCL2-associated athanogene 5 (Bag5). This complex promotes clearance of Golgi-derived vesicles through the autophagy–lysosome system [203]. Together, although the mechanism of action for LRRK2 in autophagy, CMA and the lysosome is still missing, it appears to be strongly associated with disease mechanisms.
Endophilin-A1
SH3GL2 encoding endophilin-A1 was identified in a PD risk locus by a large-scale GWAS meta-analysis [204]. Additional evidence linking endophilin with PD includes its phosphorylation by LRRK2 [185]; association with Parkin [205] and with the synucleins [137]; and its upregulation in PD brains [206]. Endophilin’s associations with PIPs are indirect and mediated through its interactions with the PIP-phosphatase, SJ1 [64, 207] and the PIP-binding protein, dynamin [64].
Endophilin CME
Endophilin is best known for its role in CME [64, 208]. However, it is also implicated in clathrin independent, ultrafast SV endocytosis [209], and exocytosis of neurosecretory vesicles [210]. Its interactions with the endocytic proteins, dynamin and SJ1 [49], facilitate neck formation in the endocytic vesicle [209] and clathrin-uncoating [65, 211]. Impairment of endophilin expression or function results in severe deficits in SV endocytosis [65, 211].
Endophilin and autophagy
Together with SJ1, endophilin belongs to the growing family of presynaptic endocytic proteins that are involved in autophagy (reviewed in [49]). Endophilin-A1 colocalizes with autophagosomes and its loss of expression interfered with the formation or transport of autophagosomes [212, 213]. An operational switch in endophilin’s function upon its phosphorylation by LRRK2 is suggested to differentiate non-phosphorylated endophilin activity in CME and membrane tubulation, while phosphorylated endophilin facilitates the formation of highly curved membranes, serving as docking sites for the autophagic factor Atg3 [212]. Thus, phosphorylation of endophilin is suggested to balance synaptic activity between endocytosis and autophagy.
Rabs
Rab GTPases include ∼70 family members in humans. They control all aspects of intracellular vesicle trafficking by acting as regulatable switches that recruit effector molecules [214, 215]. Rab GTPases are reversibly anchored into membranes by hydrophobic geranylgeranyl groups. Rabs cycle between two states, an active GTP-bound and an inactive GDP-bound state. The conversion between active and inactive states is catalyzed by guanine nucleotide exchange factors (GEFs) and GTPase-activating proteins (GAPs) [216].
Rabs and PD
Mutations in Rab39b (Park 21 locus) predispose to PD in humans [217, 218]. Additional genetic findings link Rab7L1, a LRRK2 effector protein also known as Rab29 [179, 180], with PD [219]. Overexpression of Rab1, Rab3a, Rab8a and Rab11 attenuate α-Syn-induced cytotoxicity in cell and animal models [220–222]. The potential involvement of additional Rabs is supported by data showing that LRRK2 interacts with and phosphorylates several Rab proteins, including Rab5 and Rab7 [172, 223–226]. The phosphorylation of Rab proteins is augmented by PD-associated mutations in LRRK2, which interfere with Rabs’ ability to bind both upstream regulatory proteins and downstream effector proteins [223, 224]. In addition, Vps35, a PD-associated gene [227] that is part of the retromer complex, interacts with Rab7 [228] to recruit the retromer complex to the endosomal membrane [229, 230].
Rab5, Rab7, endo-lysosomes and PIPs
The conversion between the Rab-GTP and Rab-GDP bound states, regulated by GEFs and GAPs, is mediated by the content of specific PIPs on the membrane [231, 232]. Rabs associate with PIPs, yet, these associations are generally indirect and mediated by other proteins (Table 1). Rabs regulate the recruitment and activation of PIP kinases and phosphatases [233]. For example, Rab35 binds and recruits OCRL, a PI4,5P2 5-phosphatase, to facilitate PI4,5P2 hydrolysis and clathrin-uncoating in CME, and the subsequent steps of cargo sorting through endosomes [234, 235]. A dynamic conversion of PIPs occurs following vesicle uncoating, where PI4-phosphates (e.g., PI4P and PI4,5P2 [22]) are converted to PI3P alongside with recruitment of Rab5 to the vesicle as it matures into Rab5-positive early endosome [236]. At early endosomes, Rab5 interacts with the PI 3-kinases Vps34, PI3Kβ and PI3KC2γ to synthesize PI3P [236, 237]. Rab5 also interacts with the PIP-phosphatases, type II inositol 5-phosphatase (INPP5B) [238] and Sac2/INPP5F inositol 4-phosphatase [27].
A shift in PIPs composition couples the transition between early to late endosomes and the replacement of Rab5 with Rab7 [214, 239]. Similar to Rab5, Rab7 interacts with Vps34 [240]. However, Rab7-positive endosomes are associated with higher PI4P levels, primarily produced by PI4K2A kinase [231]. Rab7 interacts with the PI4P 5-kinase, PIP5Kγ [241] and the acute conversion of endosomal PI4P to PI4,5P2 causes Rab7 to dissociate from late endosomes [231]. Recent studies suggest the occurrence of a late endosomal regulatory loop that impacts autophagosome flux and involves Rab7 cycling and PI4P to PI4,5P2 conversion [231, 241]. That is, interference with PI4,5P2 synthesis on the late endosomal membrane resulted in accumulation of LC3-positive structures with defective autophagosome-lysosome fusion.
The retromer
The retromer is a peripheral membrane protein complex that plays an instrumental role in protein recycling from endosomes to the trans-Golgi network. The retromer consists of two distinct sub-complexes, a membrane recognizing, PI3P binding sorting nexin (SNX) complex and a cargo recognition, vacuolar protein sorting (Vps) complex. Importantly, a mutation in the VPS35 gene encoding a retromer core protein, is a rare cause of familial PD [227, 242]. An involvement of the retromer and the specific D620N mutation in VPS35 has been implicated in dysfunction of the dopaminergic synapse [243] and neurodegeneration [244–248]. The D620N mutation in VPS35 was also shown to enhance LRRK2-mediated phosphorylation of several Rab members [182]. Finally, Rab7 interacts with VPS35 and recruits the retromer to late endosomes [229], in a mechanism that also requires Rab5 [249].
A role for the retromer in SV recycling, autophagy and lysosomal degradation has been reported [250, 251]. The D620N mutation in VPS35 disrupted cargo sorting [252]; associated with an abnormal endosome morphology [253]; interfered with endosomal subcellular localization and displayed evidence of retention of early and late endosome markers [252]. Moreover, the D620N mutation poorly associates with SNX3 that binds PI3P on endosomal membranes and impairs the recruitment of the WASH complex to endosomes. However, the effects of D620N mutations in autophagy are not clear yet [253, 254].
PIPs AND NON-VESICULAR TRANSPORT
Cells rely on lipid transport mechanisms to maintain structural and functional membrane plasticity, and replenish PIPs levels at the plasma membrane. Vesicular membrane trafficking is an essential route for lipid transfer between cell membranes. Following transport, PIPs and their precursor PI may serve as substrates for enzymatic conversions that will generate the required local PIP composition for membrane activities.
A key mechanism that drives local delivery of lipids from the endoplasmic reticulum (ER), the site of PI synthesis, to the plasma membrane as well as other organellar membranes, is non-vesicular lipid transport [255]. A mechanism mediated by MCS, formed between two distinct organellar membrane compartments [256, 257]. MCS are stabilized by tethering complexes that maintain close proximity between the connecting membranes but without membrane fusion. This direct route of communication is regulating lipid exchange between membranes [255]. PIPs, including PI4P and PI4,5P2, are playing fundamental roles in non-vesicular lipid transport by serving for the exchange of lipids between the plasma membrane and the ER, mediated by lipid transfer proteins [3]. A mechanistic cooperation between vesicular trafficking and non-vesicular lipid transport has recently been implicated in phagolysosome resolution, a late stage in the phagolysosomal process that facilitates the absorbance of phagolysosomal contents. Transfer of PI4P from the phagolysosomes to the ER through membrane contact sites plays a significant role in this process [258]. Moreover, de novo phospholipid synthesis at the ER was shown to support the expansion of the growing phagophore vesicle by generating stable contacts between the membranes [259]. Of interest, the PD-associated VPS13C gene encodes for a lipid transporter, acting at MCSs [242, 261]. In addition, α-Syn was detected MCSs formed between the ER and mitochondria [262].
β-GLUCOCEREBROSIDASE (GBA) AND PIPs
Although not directly related to the focus of this review, it is of interest to include a recent study that reported evidence for a role for PIPs in the cell trafficking of GBA, a sphingolipids hydrolyze [263]. Homozygous GBA mutations are a genetic cause of Gaucher disease. However, heterozygous carriers of GBA mutations have significantly increased risk for PD [264, 265]. Data show that the catalytic activity of the PI 4-kinases, PI4KIIIβ and PI4KIIα are required for the physiological trafficking of GBA along the route of its synthesis and maturation to lysosomes, where it is involved in sphingolipids catabolism.
CONCLUDING REMARKS
Understanding the implications of altered PIP homeostasis in PD mechanisms may provide clues to solve open questions and controversies over the function and dysfunction of several PD genes, improve disease diagnosis and management. Attempts are currently being made to develop specific chemical modulators for specific PIP-modifying enzymes as therapeutic strategies for diseases, including, infectious diseases, cancer and neurodegenerations. These developments hold promise for therapeutic approaches that seek the restoration of a physiologic PIP homeostasis.
Footnotes
ACKNOWLEDGMENTS
This work was funded by the Israel Science Foundation 182/12.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.