
Abstract
Evidence shows that altered retinoic acid signaling may contribute to the pathogenesis and pathophysiology of Parkinson’s disease (PD). Retinoic acid is the bioactive derivative of the lipophilic vitamin A. Vitamin A is involved in several important homeostatic processes, such as cell differentiation, antioxidant activity, inflammation and neuronal plasticity. The role of vitamin A and its derivatives in the pathogenesis and pathophysiology of neurodegenerative diseases, and their potential as therapeutics, has drawn attention for more than 10 years. However, the literature sits in disparate fields. Vitamin A could act at the crossroad of multiple environmental and genetic factors of PD. The purpose of this review is to outline what is known about the role of vitamin A metabolism in the pathogenesis and pathophysiology of PD. We examine key biological systems and mechanisms that are under the control of vitamin A and its derivatives, which are (or could be) exploited for therapeutic potential in PD: the survival of dopaminergic neurons, oxidative stress, neuroinflammation, circadian rhythms, homeostasis of the enteric nervous system, and hormonal systems. We focus on the pivotal role of ALDH1A1, an enzyme expressed by dopaminergic neurons for the detoxification of these neurons, which is under the control of retinoic acid. By providing an integrated summary, this review will guide future studies on the potential role of vitamin A in the management of symptoms, health and wellbeing for PD patients.
INTRODUCTION
Even though pharmacological treatments exist to control motor symptoms in Parkinson’s disease (PD), patients often require more holistic care in order to improve their daily quality of life and wellbeing. As such, nutrition becomes increasingly a focus for patients, medical teams and caregivers. In addition to influencing the daily management of the disease, nutrition is a potential disease-modifying factor that can either slow down the course of neurodegeneration when it is optimized, or worsen it when inadequate. Therefore, it is critical to understand what is suitable for parkinsonian patients in terms of food habits, diet, and nutrients to maximize health and wellbeing. So far, there is no consensus for an ideal diet or crucial ingredient. However, in recent years, researchers and clinicians have focused attention on candidate nutrients, such as omega-3 polyunsaturated fatty acids or vitamins. Widespread evidence, mainly in the preclinical literature, suggests that altered vitamin A signaling may contribute to the pathophysiology of PD. However, no clear consensus has been made on the underlying mechanisms.
Vitamin A is a lipophilic molecule that is exclusively provided by food. Vitamin A is the unique precursor of retinoic acid (RA), a molecule that modulates gene transcription through its binding to nuclear receptors [1]. Through its derivatives, vitamin A is involved in many important homeostatic processes in the organism, such as cell differentiation, antioxidant power, inflammation and neuronal plasticity. The role of vitamin A derivatives in the pathophysiology of neurodegenerative diseases, and their potential as therapeutics, has been carefully studied for more than 10 years, especially for Alzheimer’s disease, and to a lesser extent PD [2–8]. However, the literature sits in disparate fields, does not focus on the importance of vitamin A metabolism, and has not been concisely summarized with the view of maximizing PD patient wellbeing.
The purpose of this review is to outline what is known about the role of vitamin A metabolism in the pathogenesis and pathophysiology of PD. We examine key biological systems and mechanisms that are under the control of vitamin A and its bioactive derivative, RA, which are (or could be) exploited for therapeutic potential in PD: the survival of dopaminergic neurons in the substantia nigra pars compacta (SNc), oxidative stress, and function of immune, enteric nervous, and hormonal systems. By providing an integrated summary, this review will guide future studies on the potential role of vitamin A in the management of symptoms, health and wellbeing for PD patients.
CLINICAL AND PRECLINICAL EVIDENCE FOR A ROLE OF VITAMIN A IN THE PATHOGENESIS AND PATHOPHYSIOLOGY OF PD
Metabolism and function of vitamin A
Vitamin A (retinol) is a fat-soluble vitamin that is essential for the organism. Vitamin A is found in foods derived from animals (meat, milk, eggs) in the form of retinol esters, or in vegetable sources in the form of carotenoids that are metabolized to retinol by intestinal enzymes [9, 10]. The bioactive metabolites of vitamin A form the group of retinoid compounds or retinoids (Fig. 1). The main active metabolite is all-trans RA, called RA in this review, but other isomers also display biological activity, such as 9-cis or 13-cis-RA [9] (Fig. 1). RA acts through nuclear receptors of the steroid receptor family, namely RA receptors (RAR), which can exist in 3 different subtypes: RAR-α, RAR-β and RAR-γ [3]. The receptor subtypes differ mainly by their different locations in the body, and particularly in different brain areas [11]. RAR act as heterodimers with retinoid-X receptor (RXR) to bind DNA on RA response element (RARE) sequences located in promoter regions to activate (or repress) gene expression. RXRs also exist in 3 isoforms, RXR-α, RXR-β and RXR-γ and they function as a heterodimer partner for many other nuclear receptors [12], such as thyroid hormone receptor. The signaling of RXR is very complex and not yet fully understood. Of note, multiple ligands have been found for RXR, including retinoids, but the endogenous ligand that is most likely to activate RXR is 9-cis-dihydro RA (9CDHRA) [13]. Furthermore, RXR can also function without ligands in some cases [12–14].
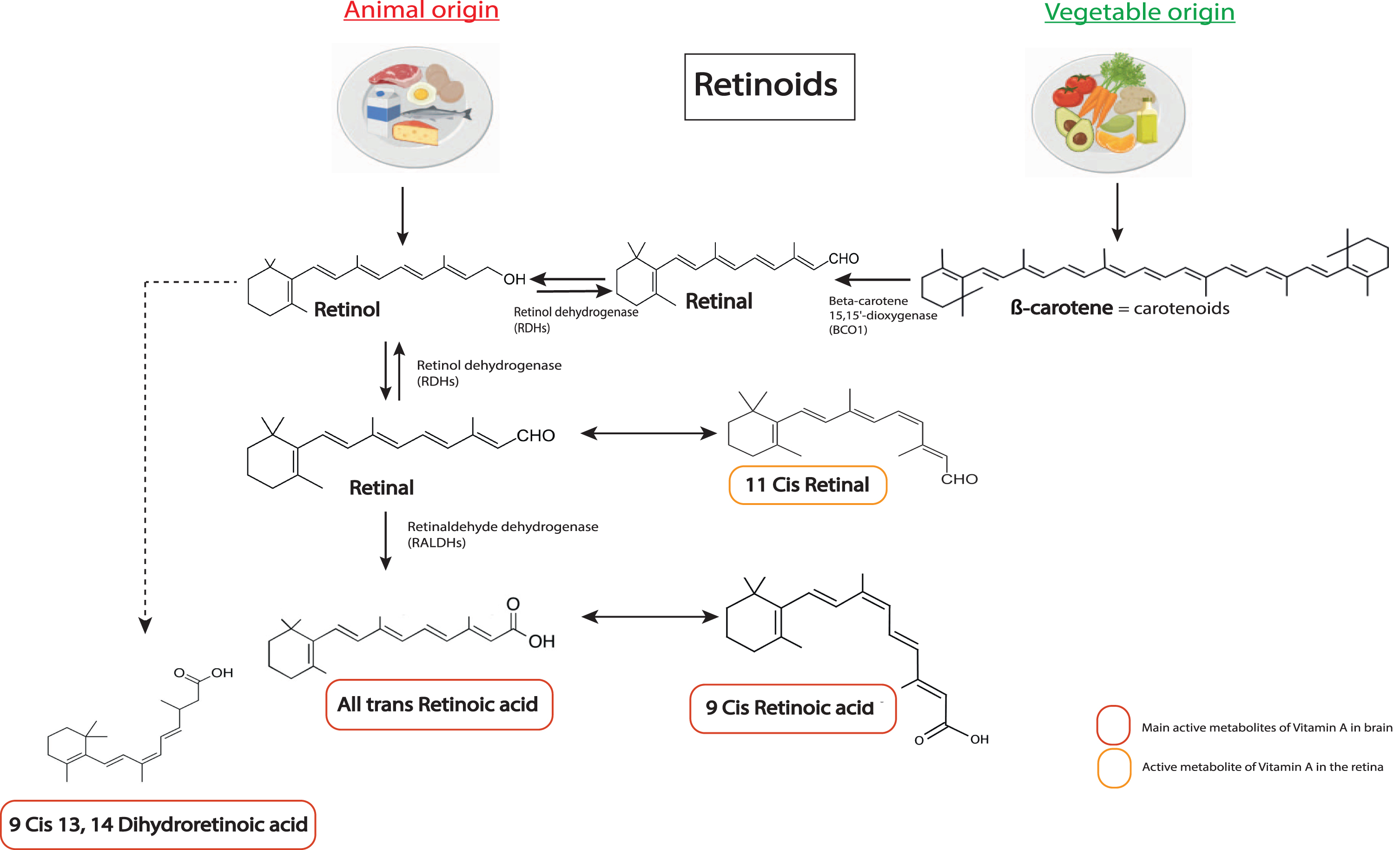
Chemical structures of the main retinoids metabolized in the body. Retinol (vitamin A) is directly provided by animal sources. Carotenoids, such as β-carotene, are precursors of retinol from vegetable sources that can be converted into retinol by the organism. Retinol is metabolized into retinal by enzymes of the RDHs family, which can also be converted back to retinol. Retinal can be metabolized into 11 cis retinal. A key bioactive metabolite produced from retinal is RA (all trans retinoic acid), which is irreversibly metabolized by a RALDH protein (ALDH1A1 belongs to RALDHs). RA can be metabolized in 11-Cis Retinal, which is an active metabolite in the retina, and in 9-cis RA, another key active metabolite for the brain. Alternatively, retinol can be transformed in 9CDHRA, an endogenous RXR ligand. RDHs, retinol dehydrogenases; RALDHs, retinaldehydes dehydrogenases.
Through its nuclear receptors (more often RAR-RXR heterodimers), RA controls the transcription of hundreds of genes, belonging to various gene pathways, including dopamine signaling and plasticity [3, 15]. Throughout life, RA is involved in cell growth, differentiation and cell death: during development, the action of RA is crucial since it is involved in the patterning of tissues, especially in the central nervous system [1, 5]; at adult age, RA continues to be involved in the maintenance and homeostasis of cells [16], and controls inflammatory responses, hormone actions, reproduction and vision [16]. The endocrine system is also tightly related to the retinoid system because RXR forms heterodimers (sometimes in an obligatory role) for some hormone nuclear receptors, such as thyroid or vitamin D receptors [17].
Vitamin A and RA are stored in liver cells, and are released on demand to act at target organs, such as the brain. Vitamin A is transformed into retinaldehyde by retinol dehydrogenase (RDH) enzymes, and retinaldehyde is further transformed in RA by retinaldehyde dehydrogenase (RALDH) enzymes [5, 10] (Fig. 2). Due to their lipophilic nature, retinoids are carried by specific proteins in the blood (retinol binding proteins, RBP) and in cells (cellular retinol binding proteins, CRBP, and cellular retinoic acid binding proteins, CRABP) to be taken to, and have action, at other organs (Fig. 2). Intriguingly, extracellular carriers (RBP) and the transmembrane transporter (STRA6 is stimulated by retinoic acid 6) have been identified for retinol, but not for RA, yet RA is clearly present in the blood [18]. In addition, STRA6 is not present on every cell, suggesting passive diffusion or another transporter, not yet identified, play a role [19].
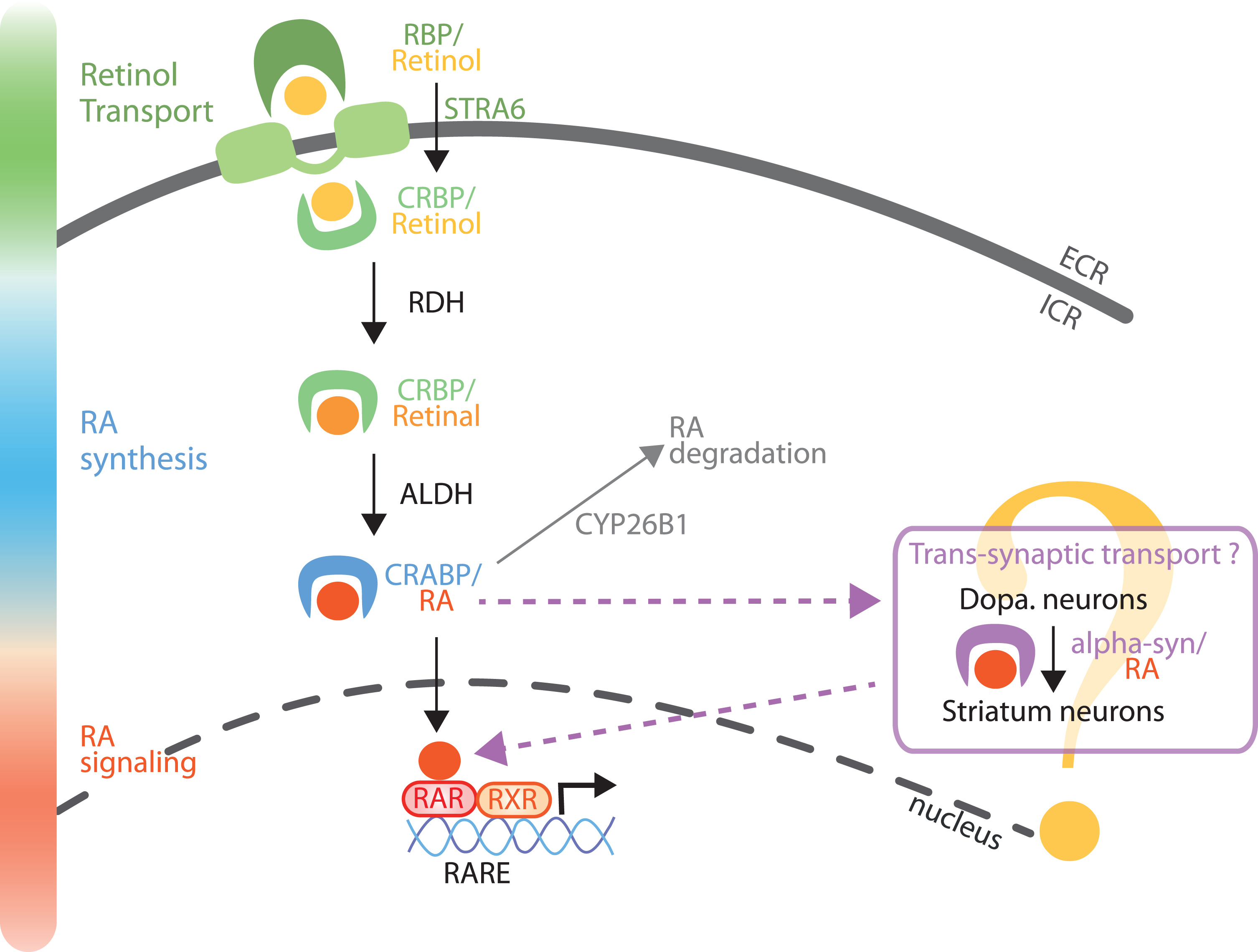
Model of RA metabolism and signaling in the nigro-striatal pathway. Retinol (vitamin A) is transported in the extracellular fluids by RBP, and internalized into the cells by STRA6. Retinol can also diffuse across the plasma membrane or through other transporters, not yet identified. Retinol is then bound to CRBP and metabolized into retinal by enzymes of the RDHs family. Retinal is further metabolized into RA by a RALDH protein (notably ALDH1A1 in a sub-population of SNc neurons). RA is then transported to the nucleus bound to CRABP protein. In the nucleus, RA binds to RA receptors (RARs), which activate the control of gene expression by RAR/RXR dimers on genes with a RARE sequence in their promoter. Furthermore, as a trans-synaptic factor, RA can travel trans-synaptically from SNc neurons to striatum neurons [24]. From the literature, it is possible that alpha-synuclein may serve as a cargo protein for the trans-synaptic transport of RA [26]. Finally, RA can be degraded by the Cyp26B1 enzyme. RBP, retinol binding proteins; STRA6, transporter stimulated by retinoic acid 6; CRBP, cellular retinol binding protein; CRABP, cellular retinoic acid binding protein; RDHs, retinol dehydrogenases; RALDHs, retinaldehyde dehydrogenases family (including ALDH1A1); RARE, retinoic acid receptor response element.
In the brain, all the machinery for retinoid metabolism/catabolism, receptors and transport are present, with a specific distribution of these proteins depending on the brain structure and cell type [5, 21] (Fig. 2). Remarkably in the hippocampus, RA has been reported to be synthetized by the meninges whereas RA has action at hippocampal neurons [20]. In the dopaminergic nigro-striatal pathway, it is likely that RA synthesis and RA action are also unevenly distributed: the enzymes for production of RA are concentrated in SNc dopaminergic neurons [3, 22], whereas RA receptors are mostly expressed in striatal neurons [11]. This is in line with the paracrine action of RA that has been described [1, 23], and recent data show that RA can act as a retrograde signaling molecule in this pathway [24]. In addition, RA signaling has been involved in the patterning of a specific dopaminergic nigrostriatal pathway [25]. Of note, a recent study reports that alpha-synuclein is a RA-carrier protein that regulates transcriptional activity of retinoids [26]. This evidence is in line with increasing literature showing that alpha-synuclein is a lipid-binding protein [27]. Yet, alpha-synuclein aggregates have been found to be expressed trans-synaptically in humans with Lewy-bodies dementia [28]. Thus, alpha-synuclein could be an extracellular carrier of RA in the nigro-striatal pathway, but this has not been fully proven yet (Fig. 2). In the study [26], RA triggers the translocation of alpha-synuclein in the nucleus in SH-SY5Y. Here, we postulate that a lack of RA could impede nuclear translocation of alpha-synuclein, thus favoring accumulation and aggregation. The balance of RA’s therapeutic and pathogenic roles needs to be elucidated.
As for other target organs, retinoid signaling is important for brain function throughout life. During development, retinoid signaling is a determinant for neuronal development, maturation and differentiation [1]. In particular, RA signaling is involved in the neurogenesis and differentiation of striatal neurons [24, 29–32]. Studies with transgenic mice demonstrate that RARβ was necessary for the differentiation of D1+ medium size spiny neurons of the striatum [29], as well as the formation of striosomes [32]. Other studies show that ALDH1A1 was necessary for the expression of μ opioid receptors (highly expressed in striosomes) [24] while RALDH3 induced the GABAergic phenotype of striatal neurons through the synthesis of RA [31]. An in vitro study also showed that DARPP-32, a marker of medium size spiny neurons, was induced by RA [30]. The late phase of dopaminergic neuron differentiation is also linked to proteins involved in RA signaling, such as ALDH1A1 or NURR1 (an orphan nuclear receptor expressed in dopaminergic neurons that dimerizes with RXR) [33–36]. Interestingly, a recent study with embryonic carcinoma tissue showed that differentiation of cells can follow striatal or dopaminergic fates, depending on the RAR subtypes that are activated [37].
In the adult brain, retinoid signaling is involved in synaptic plasticity [3, 38]. On the one hand, vitamin A deficiency or dysfunction in RAR/RXR signaling (transgenic RXR-/- and/or RAR-/- mice) alters long term plasticity in the hippocampus [20, 39–42]. On the other hand, RA controls synaptic scaling, an essential mechanism that allows homeostatic synaptic plasticity [38]. When a neuronal network is deprived of excitatory activity, the strength of synapses is increased in order to compensate for this low activity, which is called synaptic scaling. RA is a major actor of synaptic scaling, through various processes depending on the system studied, such as externalization of AMPA receptors or internalization of GABAA receptors [43–49]. Of note, these mechanisms can involve non-genomic action of RA and their receptors by stimulating local dendritic protein synthesis [38, 50]. These mechanisms have been described in cell cultures or in the hippocampus, but rarely in the basal ganglia. One recent study showed that RA regulates levels of AMPA receptors in cultured nucleus accumbens neurons [51].
With aging, retinoid signaling remains necessary for brain function, however, aging processes and senescence eventually reduces vitamin A function and efficacy. Indeed, data show that the liver reduces its ability to release vitamin A and RA with age, which can lead to decreased bioavailability for target organs, especially the brain [18, 53]. This leads to reduced neurogenesis and reduced dendritic tree arborization for pyramidal neurons in the hippocampus [18, 54]. Thus, the decreased bioavailability of vitamin A with age could contribute to age-related cognitive decline, and the development of neurodegenerative processes that underlie Alzheimer’s disease and PD. Interestingly, increasing dietary vitamin A intake can prevent the decrease of vitamin A bioavailability [18, 54]. Indeed, nutritional studies show that dietary vitamin A is stored in the liver and a proportion of vitamin A is directly transported to the brain through the portal vein system [55–57]. A recent study showed that this ‘secondary pathway’ for vitamin A absorption is particularly dedicated to the brain [57]. Moreover, vitamin A supplementation increases the amount of vitamin A absorption through this pathway, favoring brain supply and ‘escaping’ or by-passing liver storage [57]. Therefore, vitamin A supplementation may be a therapeutic lever to improve bioavailability of vitamin A, and thus retinoid signaling, in the brain.
Vitamin A and PD: Clinical studies
Epidemiological and clinical studies investigating the link between vitamin A or retinoids/carotenoids and PD have not shown a clear effect (Table 1). Original studies, meta-analyses and reviews have mainly found an absence of link between the disease and the dietary intake [58–66]. More recent studies found an association between low dietary intake of β-carotene, or low levels of blood α-carotene, β-carotene and lycopene with the progression of PD [67, 68]. These studies are based on nutritional intake or retinol blood levels, and therefore they do not provide information about the vitamin A status of patients, i.e., the bioavailability of vitamin A and RA for target organs, such as the brain. The level of retinol in the blood is correlated to nutritional intake, but is also tightly controlled by homeostatic processes to maintain a constant value around 2μg/ml [52]. Thus, the retinol level in the blood is not a reliable marker of vitamin A and RA functionality. More relevant measures would be gene expression of RA receptors (RAR/RXR) or retinol level in the cerebral spinal fluid [69]. Measuring RA concentration in the blood or the cerebral spinal fluid would also be more informative, but it is highly challenging due to its biochemical instability [70].
Summary of the main clinical studies that have directly or indirectly investigated the relationship between vitamin A and Parkinson’s disease
*compared to control cases.
Of note, ALDH1A1, a synthetic enzyme for RA, has been described as a biomarker for PD, since its reduced expression has been found in the blood and brain of PD patients [71–73] (see below).
Vitamin A and PD: Preclinical studies
Despite the lack of association between dietary vitamin A and PD in humans, important links have been established between the pathophysiology of the disease and proteins involved in the metabolism of vitamin A and retinoids. Preclinical evidence for vitamin A’s role in the pathogenesis of PD has been established by various manipulations of vitamin A and its pathways: dietary supplementation, dietary deficiency, knock-out mice for retinoid receptors, treatments with vitamin A derivatives in vivo or in vitro.
First, evidence came from the discovery that the development and fate of the nigro-striatal pathway is under the control of nuclear receptors from the retinoid receptor family, namely RAR, RXR and Nurr1 [8, 75]. Of note, the striatum is the brain structure that expresses the most RARβ and RXRγ [11] and expression of D1 and D2 receptors in the striatum is under the control of retinoid receptors [74, 76]. Furthermore, Nurr1 is an orphan nuclear receptor expressed in dopaminergic neurons that dimerize with RXR and has been linked to PD [75]. In mutant mice lacking RAR/RXR or Nurr1, dopaminergic neuron development was altered and associated with motor impairments in adult age [8, 76]. These early observations indicate that retinoid receptors are involved in the development of the nigro-striatal pathway, with consequences on motor function, and it has been postulated that retinoids could be involved in the pathogenesis of PD (Table 2).
Summary of the main studies relating the impact of vitamin A pathway alterations (dietary intake, receptors or ALDH1A1) in Parkinson’s disease rodent models. VAD, vitamin A deficient; D1R, dopamine D1 receptor; D2R, dopamine D2 receptor; TH, tyrosine hydroxylase; DA, dopamine; DOPAC, 3,4-dihydroxyphenylacetic acid; 5HT, serotonine; HVA, homovanillic acid: A53T mice, transgenic mice that overexpress human α-synuclein with a PD-associated mutation (A53T)
*compared to control group.
Later, important results have been obtained with the ligand of these receptors, namely RA or synthetic derivatives (Table 3). Injection of nanoparticles loaded with RA powerfully reduces dopamine neuron degeneration in the SNc in a mouse model of PD [77]. Similarly, pre-treatment with 9-cis RA reduces midbrain dopamine neuron loss in a rat model of PD [78], as well as neurotoxicity induced by methamphetamine [79]. Furthermore, protection of midbrain dopamine neurons is afforded by exogenous ligand activation of retinoid-related receptors (Nurr1-RXR heterodimers) [80, 81]. In particular, chronic in vivo injections of Nurr1:RXRα heterodimer activators in mouse models of PD reduce motor impairments, increase TH+ neurons in the SNc and TH+ terminals in the striatum [80]. Vitamin A has also been shown, in vitro, to destabilize aggregates of α-synuclein [82], although a contrary result has been reported [83].
Summary of the main studies relating the impact of vitamin A pathway enhancement (dietary intake, vitamin A derivatives, or receptors ligands) in Parkinson’s disease rodent models
*compared to PD model without supplementation.
Beyond the use of retinoid derivatives, some studies have directly investigated the role of dietary vitamin A in the pathogenesis and pathophysiology of PD (Tables 23). Vitamin A deficient rats have motor impairments similar to those observed in rat models of PD or in mice lacking retinoid receptors [84]. Another study reported that vitamin A deficiency impaired dopaminergic transmission [85]. Furthermore, the only study that has investigated the impact of dietary vitamin A supplementation showed that vitamin A administration for 4 weeks before the 6-OHDA lesion in the SNc improved movement deficits in 6-OHDA rats [86]. While the behavioural improvement was small, the effect on neuronal survival was not clear. Therefore, more studies are needed with vitamin A supplementation in animal models.
From these preclinical studies, it is clear that vitamin A derivatives and their receptors are involved in the homeostasis of SNc dopaminergic neurons. However, direct involvement of vitamin A metabolism in the pathogenesis of PD has not been established.
VITAMIN A AND DERIVATIVES FOR THE PROTECTION OF DOPAMINERGIC NEURONS IN THE BRAIN
Antioxidant power of vitamin A
Oxidative stress is defined as ‘an imbalance between the oxidants and antioxidants in favor of the oxidants, leading to a disruption of redox signaling and control and/or molecular damage’ [87, 88] and is a factor for neuronal degeneration in PD [89]. In this context, oxidative stress refers mainly to the formation of free radicals, reactive oxygen species (ROS) that are overproduced by complex I and II of mitochondria in dopaminergic neurons [90]. These free radicals are toxic for cells due to their reactivity and ability to damage biochemical elements. In physiological conditions, the balance between oxidants and antioxidants is maintained by enzymatic antioxidants and non-enzymatic oxidants, and retinoids are part of the antioxidant system [88]. In parkinsonism, accumulation of excessive free radicals in dopaminergic neurons underlies dysfunction of mitochondrial complex I [90, 91]. The origin of this dysfunction is not known and likely to be multiple. Yet, some features of midbrain dopaminergic neurons are thought to favor oxidative stress: (i) their highly branched unmyelinated axons have high bioenergetic demands [92], (ii) pacemaker activity of these neurons (2–10 Hz) produces a constant relatively high level of intracellular calcium level and thus causes a continuous bioenergetic burden for calcium buffering [93, 93], and (iii) metabolism of dopamine creates ROS metabolites that interfere with the function of mitochondrial complex I [90, 93]. Of note, dopamine-3,4-dihydroxyphenylacetaldehyde (DOPAL) is a highly reactive metabolite of dopamine that exacerbates α-synuclein aggregation [94]. These features are not exhaustive and other pro-oxidant factors in dopaminergic neurons, such as metals, do exist (for review, see [89]). Together, multiple sources of free radical production are proposed to explain the vulnerability of midbrain dopaminergic neurons to degenerate in PD.
Retinoids and carotenoids are antioxidants due to their ability to inhibit peroxidation, scavenge free radicals and maintain the balance between oxidants and antioxidants [91, 95–98]. However, studies indicate that high doses of vitamin A supplementation can conversely induce an imbalance between oxidants and pro-oxidants, in favor of oxidants [99–101]. Most of the data concerning the antioxidant role of vitamin A and associated retinoids have been collected from the heart or liver, but data have rarely explored the antioxidant (or pro-oxidant) role of vitamin A in the neurodegeneration of dopaminergic neurons. Indeed, the role of antioxidants in the progression of PD has been investigated for other vitamins, such as vitamin E or vitamin C [91], but not directly for vitamin A or other retinoids. Therefore, the role of dietary vitamin A on oxidative stress in PD remains to be explicitly studied.
The neuroinflammation action of vitamin A
Neuroinflammation is a common feature of neurodegenerative diseases. Even though the role of neuroinflammation in the pathophysiology of these diseases is established [102], involvement of neuroinflammation in their pathogenesis is not yet demonstrated. However, reducing neuroinflammation is generally a way to curtail disease progression and several pharmacological agents have been tested to address this gap in understanding.
In this context, it is logical to investigate the potential anti-inflammatory role of vitamin A in PD. Indeed, vitamin A is involved in the development, maturation and differentiation of immune organs and cells [103]. Moreover, vitamin A has the propensity to reduce pro-inflammatory factors and enhance anti-inflammatory factors, in the periphery as well as in the brain [104, 105]. Specific to neuroinflammation, in vitro studies demonstrate that application of RA to astrocytes or microglial cells reduces inflammatory responses induced by lipopolysaccharide (LPS), a bacterial endotoxin [106, 107]. Related to PD, a study clearly demonstrated that pharmacological stimulation of RAR in vitro and in vivo reduces neurodegeneration of midbrain dopaminergic neurons induced by an immune challenge. A more recent study observed a protective effect of dietary vitamin A in a rat model of PD by reducing pro-inflammatory factors (TNF-α, IL-1β, Iba-1), which was accompanied with an increase in GFAP staining, suggesting increased astrocytic reactivity [86].
The role of retinoids in dampening neuroinflammation appears an important mechanism to impede neurodegeneration in PD. However, given the limited number of preclinical studies, more research is needed to better understand the underlying mechanisms.
In PD, the classical scheme of neuroinflammation indicates that pro-inflammatory factors are produced by activated microglia and astrocytes. In addition, pro-inflammatory factors released by peripheral macrophages can penetrate brain tissue because the blood-brain barrier becomes leaky [108]. It is likely that these pro-inflammatory factors contribute to oxidative stress in dopaminergic neurons, which further activate neuroinflammatory processes [60, 109].
Related to neuroinflammation and oxidative stress, studies exploring the role of vitamin A metabolism in the control of circadian rhythms and sleep deserve attention. A main prodromal symptom observed in PD is sleep disorder [110] and it is also observed in rat models of PD [111]. These sleep disorders are mainly a lack of REM sleep, but can also exhibit other forms of sleep deficits. Retinoid signaling is involved in sleep wave rhythms, as demonstrated in mice with RAR KO mice [112], vitamin A deficiency [85] or pharmacological inhibitors [113]. Importantly, these studies also highlight that reduced vitamin A function induced changes in sleep wave rhythms that were associated with alterations in striato-nigral dopaminergic transmission [85, 113].
In addition, circadian rhythms are also altered in PD, which could contribute (even initiate) to the vicious cycle between neuroinflammation and oxidative stress [114]. Multiple evidence shows that vitamin A, through RA and RAR/RXR receptors, controls the gene of the circadian oscillator [115].
Vitamin A and neurogenesis
Under physiological conditions, neurogenesis happens in restricted regions of the adult mammalian brain such as the subgranular zone of hippocampal dentate gyrus and the subventricular zone (SVZ) of lateral ventricles, two areas that neural stem cells develop [116]. Neurogenic niches are endogenous sources for new neurons that can be used for brain repair strategies [117]. It has been demonstrated that RA is necessary for adult hippocampal neurogenesis [3, 118]. Indeed, nutritional vitamin A deficiency induced spatial memory deficits and adult hippocampal neurogenesis alterations, that are corrected by RA treatment or vitamin A supplementation in rodents [119, 120].
In rodent models of PD, due to the proximity of the SVZ with the striatum (and thus dopaminergic afferences), stem cells from this area can be recruited following a lesion. Some studies have established the potential therapeutic benefit of neurotrophic factors and proposed them as suitable candidates for stimulating neurogenesis after induction of neurodegeneration [121]. Among these neurotrophic factors, CDNF (cerebral dopamine neurotrophic factor) is the most promising treatment for PD due to its selective effects on dopaminergic neurons. Thus, it has been shown that intra-SVZ administration of CDNF enhances the proliferation and migration of neural stem cells toward the 6-hydroxydopamine (6-OHDA)-lesioned striatum accompanied by improvement of motor dysfunctions in parkinsonian rats [122].
In this context, vitamin A and RA are also promising molecules to enhance the generation and long-term survival of SVZ-derived neurons after PD lesions. Indeed, RA is a potent mitogen for SVZ neuroblasts, and is required for their migration to the olfactory bulb [123]. More recently, the manipulation of endogenous stem cell populations from the SVZ created an opportunity to induce neurogenesis and influence brain regenerative capacities in the adult brain. Herein, new approaches demonstrated the ability of RA loaded-nanoparticles to induce neurogenesis exclusively after being internalized by SVZ stem cells both in vivo and in vitro. Similarly, combined RA treatment with environmental enrichment enhanced the generation and long-term survival of SVZ-derived striatal neurons after stroke [127].
These data indicate that recruiting endogenous SVZ neural stem cells toward striatal regions exhibiting retrograde degeneration of dopaminergic afferents in PD brains by stimulating retinoid signaling could be a way to slow down the neurodegeneration of nigrostriatal dopaminergic neurons, and to allow functional recovery. Retinoids could also be as used a molecular factor to improve the durability of cell grafts that are currently developed to promote cell repair [4, 128]. In this later case, retinoid signaling could be envisaged as a treatment strategy.
Pivotal role for ALDH1A1
Beside the potential role for vitamin A to control neuroinflammation and oxidative stress, vitamin A’s action may also be specific to dopaminergic neurons by controlling the retinaldehyde dehydrogenase 1 (ALDH1A1) enzyme.
As mentioned above, RA in the adult brain controls homeostasis of dopaminergic neurons and dopaminergic transmission by controlling the expression of tyrosine hydroxylase, D2-like receptors, and the RA synthetic enzyme, ALDH1A1 [3]. ALDH1A1 is a retinaldehyde dehydrogenase enzyme that is part of the aldehyde dehydrogenase superfamily of enzymes whose main function is to synthesize RA from retinal [129] (Figs. 12). These enzymes form a vast family that has multiple functions, such as acetaldehyde detoxification [129]. It exists as 3 isotypes of RALDH: RALDH1 (ALDH1A1), RALDH2 and RALDH3. ALDH1A1 is coded by the gene Aldh1a1 but the enzyme has multiple names in the literature: Ald1a1, RALDH1, ALDH1, Ahd2. Excluding dopaminergic neurons, RALDH2 is the main isotype expressed in the brain [130]. Dopaminergic neurons are unique in the brain because they are the only ones to express the ALDH1A1 isotype in the adult brain [21]. Specifically, only a subset of midbrain dopaminergic neurons in the SNc and ventral tegmental area express this enzyme [131], making ALDH1A1 a marker for this subpopulation. This dopaminergic subpopulation is interconnected with striatal neurons and mice lacking these neurons display motor impairments [132]. Mice lacking the ALDH1A1 enzyme also display motor impairments and alterations in dopamine release [132–134], consistent with associated motor deficits and reduced levels of ALDH1A1 reported from clinical studies [71–73] (Table 1). Remarkably, ALDH1A1-/- mice also display a lack of mu opioid receptors MOR1 expression in striosomes compartments [24, 25].
In addition to these preclinical data brought by ALDH1A1-/- mice, it appears that ALDH1A1 labelled neurons correspond to the dopaminergic subpopulation that is the most vulnerable to degeneration in PD [135, 136]. Specifically, data obtained from humans and rodent models of PD indicate that neurons that degenerate early are those that express the gene Aldh1a1 coding for ALDH1A1 [73, 137].
This raises the question of why ALDH1A1 expressing neurons are more vulnerable in PD? Is ALDH1A1 a marker for neuroprotection or neurodegeneration? ALDH1A1 is neuroprotective, because these dopaminergic neurons stop expressing ALDH1A1 before degenerating [71, 137]. Importantly, these observations are not only based on animal studies, since a reduction of ALDH1A1+ neurons has also been observed in the SNc of patients with PD [137]. Therefore, the scenario envisaged is that ALDH1A1+ neurons stop expressing ALDH1A1 (for unknown reasons, discussed later), which triggers their degeneration. In support with this, accumulating data show that ALDH1A1, beyond synthesizing RA, plays an essential detoxifying role. Indeed, ALDH1A1 has the ability to degrade DOPAL, a highly oxidizing dopamine metabolite that induces oxidative stress and aggregation of α-synuclein proteins [94]. Thus, DOPAL could be a major contributor to dopaminergic neuron loss in PD, whereas ALDH1A1 would preserve dopamine neurons by degrading DOPAL [73, 138]. This potential mechanism is consistent with the ‘catecholaldehyde hypothesis’ [139–141].
In support of the catecholaldehyde hypothesis, unintentional over-inhibition of ALDH1A in humans has produced patients with parkinsonism symptoms, following disulfiram (used as Antabuse® for the treatment of alcoholism) intoxication [142, 143]. Furthermore, the toxicity of some pesticides, such as benomyl or rotenone, is due to the inhibition of aldehyde dehydrogenase enzymes [138, 144–146]. However, in these articles, the ALDHs that are studied are mostly mitochondrial ones and ALDH1A1 is not investigated. Still, pesticide exposure is a known cause of PD and these studies highlight the crucial role of ALDH enzymes in the survival of dopaminergic neurons.
Beyond this pharmacological/exogenous inhibition of ALDH1A1, we do not know why and how dopaminergic neurons stop expressing ALDH1A1 in parkinsonian patients. Nevertheless, ALDH1A1 expression is controlled by retinoid signaling. Knowing that ALDH1A1 expression may be an important prodromal biomarker for PD [73, 137], we propose the novel hypothesis that vitamin A metabolism is involved in PD pathogenesis. Indeed, ALDH1A1 expression is modulated by RA levels. More precisely, RA stimulates the expression of PITX3, a transcription factor that controls expression of ALDH1A1 [77, 148]. Mice deficient for PITX3 exhibit strong dopamine loss in the SNc [149–151] and exhibit motor symptoms that are similar to animals with neurotoxic lesions [152, 153], which can be reversed by RA administration [147].
In support of our hypothesis that vitamin A metabolism is involved in PD pathogenesis, we recently showed that aging in rats is accompanied by a decreased bioavailability of RA in the brain [18]. Since aging is the greatest risk factor for PD, the collapse of retinoid signaling in the brain could be a contributing factor in a vicious cycle leading to degeneration of ALDH1A1 + dopaminergic neurons in the SNc. This proposed mechanism is presented in Fig. 3. Multiple, disparate evidence supports our hypothesis. However, the real association between vitamin A status, RA bioavailability and dopaminergic neuron loss in PD remains to be demonstrated. This proposed mechanism also raises further questions about the subpopulation of dopaminergic neurons that do not express ALDH1A1. Do dopamine neurons without ALDH1A1 produce DOPAL? If yes, what are the enzymes responsible for DOPAL degradation in these neurons? What enzyme isotypes are expressed? Furthermore, data indicate that ALDH1A1 determines the GABAergic nature of neurons from the SNc [31, 154], which is in contrast (but not necessarily in contradiction) with data indicating that RA is involved in the development of dopaminergic pathways [8, 75].
In conclusion, data support the theory that ALDH1A1 is a pivotal enzyme for understanding the role of vitamin A in the neurodegeneration of dopaminergic neurons, but further investigations are needed to confirm and strengthen these data.
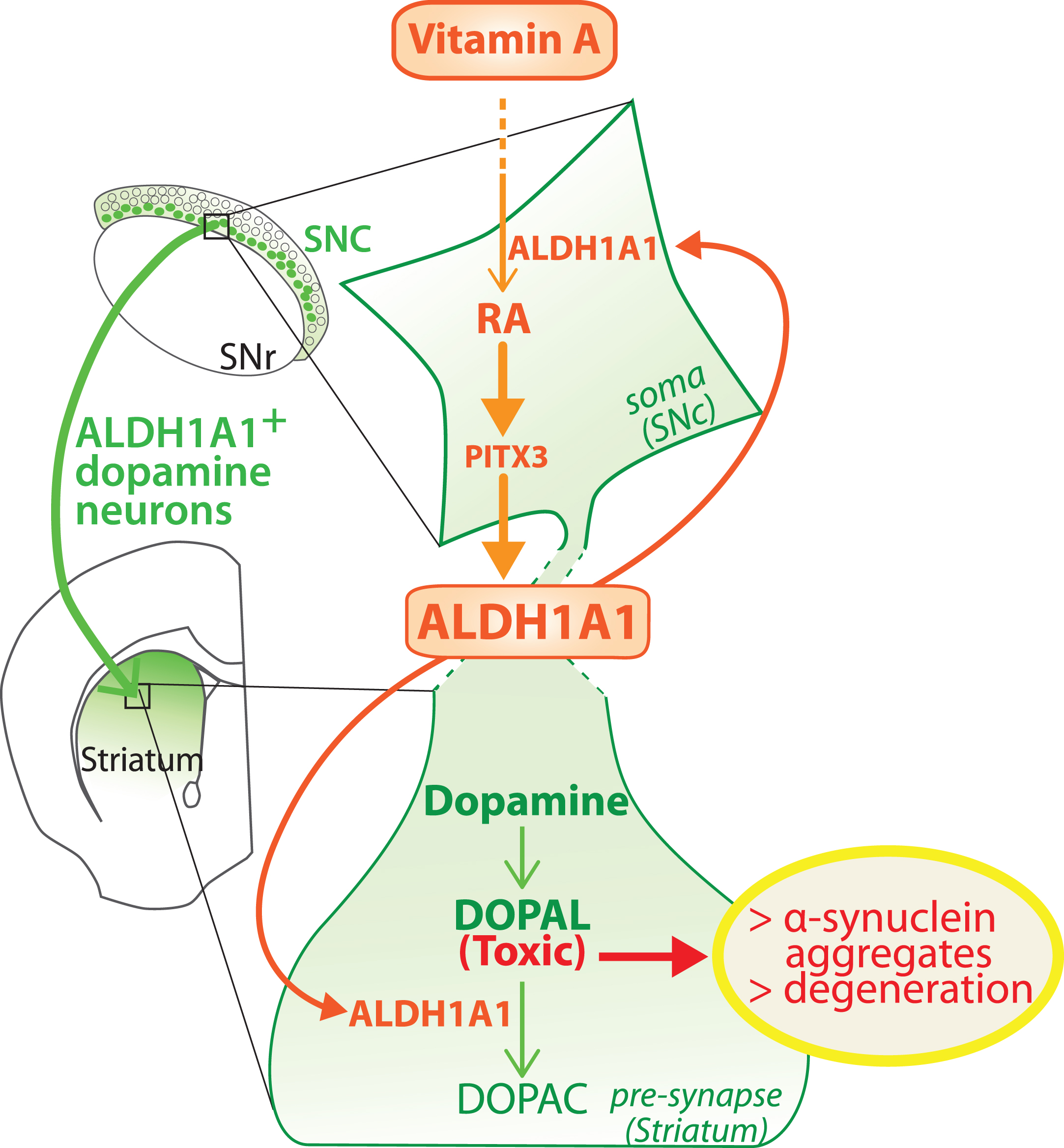
Model of the dual role played by ALDH1A1 in the nigro-striatal pathway. ALDH1A1 is involved in the metabolic pathway of RA because it synthesizes RA from retinal. In parallel, ALDH1A1 is involved in catabolic pathway of dopamine because it degrades DOPAL to DOPAC. Considering that RA controls the expression of ALDH1A1 through PITX3, the model proposes that ALDH1A1 expression is controlled by vitamin A bioavailability. SNc, substantia nigra pars compacta; SNR, substantia nigra pars reticulata.
PERIPHERAL ACTIONS OF VITAMIN A IN PD
The enteric nervous system (ENS)
The ENS is the autonomous nervous system of the gastrointestinal (GI) tract and it is connected to autonomous and central nervous systems by the vagus nerve. ENS neurons are remarkable in their diversity, and they are highly organized [155]. In particular, the ENS is rich in dopaminergic neurons.
GI dysfunctions, such as constipation or slowed gastric emptying, are symptoms frequently observed in PD, which can appear years before motor symptoms [156, 157]. These prodromal symptoms are thought to be related to altered ENS function. Since Lewy bodies and alpha-synuclein aggregates have been found in the ENS and dorsal motor nucleus of the vagus nerve in PD patients, the disease may originate from enteric alpha-synucleopathy and reach midbrain dopamine neurons through the dorsal motor nucleus of the vagus nerve [158]. These mechanisms may involve dopaminergic neurons and/or epithelial cells that produce dopamine, for the same reasons outlined earlier for CNS dopaminergic neurons. Since proposing this hypothesis in 2003, numerous preclinical and clinical studies have been conducted [159–164]. In rodents, alpha-synuclein transport from the GI to the midbrain and associated motor impairments have recently been demonstrated [157, 165]. In humans, some evidence highlights the link between GI dysfunction, synucleopathy in the ENS and severity of PD symptoms [157, 164]. However, this evidence has also been rebutted by other studies [166–169]. Therefore, the precise role of ENS synucleopathy in the pathophysiology of PD is still under debate.
In this context, we wonder whether vitamin A can protect ENS neurons via similar mechanisms proposed for midbrain dopaminergic neurons. RA plays a critical role in the development and differentiation of ENS neurons [170]. As for central dopaminergic neurons, we could extrapolate that factors involved in the development of ENS neurons are also critical for their survival at adult age. In addition, vitamin A deficiency in rats reduces the number of cholinergic and nitrergic neurons in the ENS, which is associated with altered colon motility [171]. Interestingly, reduced colon contractility has also been found in RALDH KO mice [172]. However, the impact of vitamin A deficiency on ENS dopaminergic neurons does not appear to have been investigated. From these data, we can postulate that vitamin A metabolism is important for survival of ENS dopaminergic neurons, and may protect from their degeneration, but more research is needed.
The gut microbiome
Gut microbiota composition has recently become a focus for PD researchers [173–176]. A key study in 2016 demonstrated that human gut microbiota from Parkinson’s patients worsens motor symptoms in a mouse model of PD [177]. Furthermore, a two-year follow-up study revealed that low counts of specific bacteria such as Bifidobacterium (Actinobacterium phylum) and B. fragilis (Bacteriodetes phylum) at the beginning of the study was associated with much worse symptoms of the disease two years later [178]. However, there is still a lack of preclinical and clinical data explaining the links between impoverished gut microbiome and progression of PD [179, 180].
Vitamin A deficiency may alter gut microbiota. In mice, vitamin A deficiency reduces bacteria from the Bacteroidetes phylum (to whom B. fragilis belongs to), which altered energy homeostasis of the animal overall, and resulted in glucose and insulin intolerance [181, 182]. Young rats from vitamin A deficient mothers also displayed a dysbiosis of colonic mucosal microbiota, in particular with reduced members of the Bacteriodetes phylum [183]. Another study revealed that retinol, but not other retinoids such as RA or beta-carotene, inhibits growth of B. vulgatus [184]. As a consequence, vitamin A deficiency in mice increases the growth of B. vulgatus, but the consequence of this imbalance was not evaluated. Conversely, RA is needed for Bifidobacterium growth [185].
Thus, two observations may converge; vitamin A likely modulates gut microbiota composition, and PD is associated with specific microbiota modifications. However, the lack of studies about microbiota related to PD, as well as related to vitamin A is too significant to draw any hypothesis about the mechanisms linking vitamin A dysfunction to microbiota changes in PD.
Link between vitamin A and other hormones through RXR dimerization
Finally, vitamin A may influence the development of PD pathophysiology indirectly via hormonal systems. Indeed, RXRs have a particular role because they are dimer partners (sometimes obligatory) for multiple nuclear receptors, such as thyroid receptor (TR), Nurr1, Nur77 or vitamin D receptor (VDR). Thus, retinoids (9CDHRA in particular) through their binding to RXRs are at a crossroad between multiple hormonal pathways, and vitamin A is considered a hormone [186]. Furthermore, expression of RXRs themselves are controlled by retinoids, therefore, vitamin A status can regulate the function of RXRs [187, 188]. In the context of PD, the bioavailability of vitamin A can thus have an impact on hormonal systems, which affect symptom progression, health and wellbeing, but this interaction could be easily overlooked.
The case of thyroid hormones
Thyroid receptors (TRs) are nuclear hormone receptors for which heterodimerization with RXR is obligatory. More precisely, RXR modulates the action of TR with the outcome dependent on the ligands of both receptors [17]. Vitamin A deficiency is associated with altered thyroid function, while hypothyroidism is associated to decreased RAR in humans [189]. RA modulates the expression of proteins involved in thyrocyte function and can stimulate their differentiation [189]. Vitamin A and thyroid metabolisms are thus intricately linked even though underlying mechanisms are not fully understood. With aging, levels of both RARs and TRs are decreased as shown in rats and humans [53, 190–193]. However, studies diverge on RA’s ability to restore TR mRNA levels in aged rats [53, 190–192]. Furthermore, thyroid hormones stimulate metabolism and can increase the production of ROS by mitochondria [194]. Thus, thyroid dysfunction may be involved in idiopathic PD pathophysiology through oxidative stress. In a preclinical study, increased levels of T3 have been found in 6-OHDA lesioned rats suggesting that central dopaminergic denervation can act on thyroid levels at the periphery, possibly through dysfunction of hypothalamic-pituitary axis [195]. In patients, thyroid hormone function has been poorly documented so far and results are mitigated. One study indicated that subclinical hypothyroidism was more frequent in PD patients [196]. Another study indicated an association between the severity of motor symptoms and level of free circulating thyroid hormones (fT3) [197]. However, hypothyroidism did not differ significantly between parkinsonian patients and controls in a third study [198]. Therefore, due to the lack of studies the interaction between vitamin A and thyroid function in the pathophysiology of PD has not been established. Further investigations should be conducted, particularly in patients with idiopathic origin, who are over 60 years of age at the time of diagnosis, an age where the thyroid function can be impaired by aging processes.
The case of vitamin D
Vitamin D is renowned for its roles in immune function and maintaining bone density (through the control of calcium serum levels). As for TRs, expression of vitamin D nuclear receptors (VDRs) is controlled by RA, and RXR is an obligatory heterodimer for VDRs [199]. A correlation between vitamin D levels and PD has been observed, but vitamin D supplementation has not significantly reduced disease status [200–202]. The SNc and the striatum are two structures highly enriched for VDR and 1α-hydroxylase, the enzyme that synthesizes the active form of vitamin D [203]. Pre-clinical data show that vitamin D protects dopaminergic neurons from oxidative stress [199, 203–206]. In vitro, low (but not high) doses of the active form of vitamin D protects dopaminergic neurons (primary mesencephalic cultures) from toxins [204]. In vivo, treatment with the active form of vitamin D 1 week before 6-OHDA lesion in rats increases motor activity, dopamine content in the striatum and number of TH+ neurons [205, 206]. These studies suggest that the positive effect of vitamin D is mediated by its antioxidant effect. Thus, beyond the interactions that may exist between vitamins A and D in the immune system, these two vitamins may act synergistically in the brain. However, the combined action of vitamins D and A on the survival of dopaminergic neurons in PD, notably through their partner nuclear receptors, has not been investigated. Yet, this constitutes an interesting avenue with easily translatable therapeutic outcome.
CONCLUSION
This review explored multiple mechanisms that vitamin A metabolism may contribute to PD pathogenesis and PD pathophysiology. Indeed, altered vitamin A metabolism and bioavailability is likely to contribute to oxidative stress, neuroinflammation, dopaminergic cell death, disturbance in biological rhythms and endocrine homeostasis (Fig. 4). Thus, vitamin A, as a nutritional factor may be at the crossroad of multiple environmental and genetic factors of PD. Still, the central underlying role that decreased vitamin A metabolism may have in PD has been largely ignored. This may be due to the lack of adapted methods for quantifying vitamin A metabolism and bioavailability in humans.
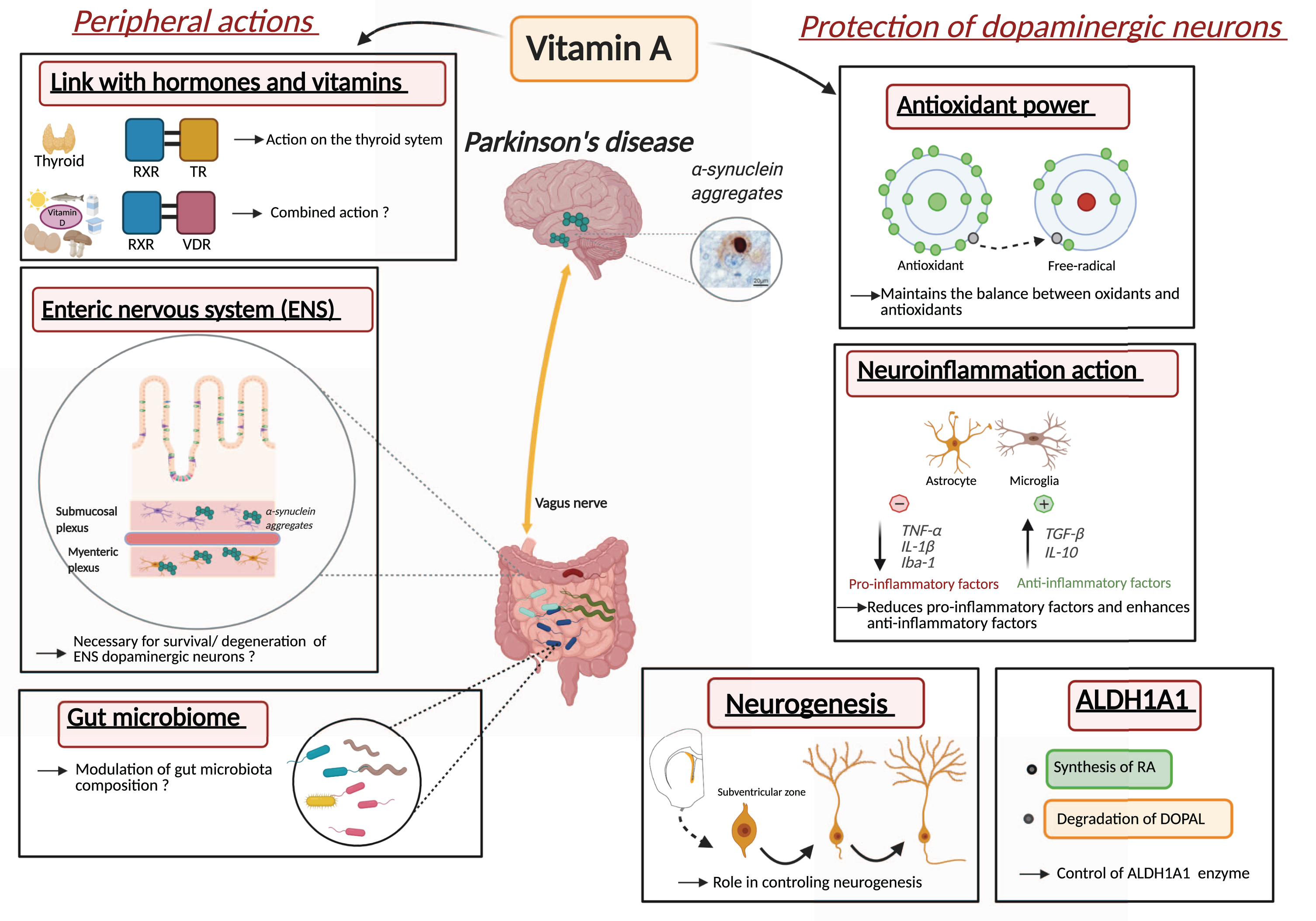
Proposed roles and mechanisms of vitamin A metabolism in the pathophysiology of Parkinson’s disease. Inset with α-synuclein aggregates from [110].
From our overview, we propose that vitamin A metabolism may be involved in the pathogenesis and pathophysiology of PD in multiple ways (Fig. 4). Of note, other possible ways have not been discussed here, such as the potential role of retinoids in autophagy. At this stage, the most promising way by which vitamin A metabolism may influence PD pathogenesis and treatment is through the impact of vitamin A on ALDH1A1 expression and neuroinflammation. A first step is to understand the role of ALDH1A1 in controlling dopaminergic cell survival, within the schema of the catecholaldehyde hypothesis. However, other mechanisms of vitamin A metabolism are likely relevant including oxidative stress, neurogenesis in the SVZ, ENS function and microbiota, thyroid hormone and vitamin D function, but more data are needed to fully understand the role of individual or combined mechanisms.
Finally, it is important to keep in mind that pharmacological treatments with retinoids have been studied for a long time and these drugs have to be used in precise conditions to preclude side effects [7]. In this framework, targeting vitamin A, and not RA and its receptors, may be a more conservative strategy since vitamin A supplementation increases bioavailability for the brain, and is better managed by organisms than derivatives, with fewer side effects. Together, a better understanding of the role played by vitamin A metabolism in PD could open the way for new approaches to dampen symptoms and improve health and wellbeing for PD patients.
Footnotes
ACKNOWLEDGMENTS
This work was supported by funding from France Parkinson, Health Research Council (17–284) and Brain Research New Zealand
CONFLICT OF INTEREST
The authors have no conflict of interest to report.