
Abstract
Background:
Genetic studies have indicated that variants in several lysosomal genes are risk factors for idiopathic Parkinson’s disease (PD). However, the role of lysosomal genes in PD in Asian populations is largely unknown.
Objective:
This study aimed to analyze rare variants in lysosomal related genes in Chinese population with early-onset and familial PD.
Methods:
In total, 1,136 participants, including 536 and 600 patients with sporadic early-onset PD (SEOPD) and familial PD, respectively, underwent whole-exome sequencing to assess the genetic etiology. Rare variants in PD were investigated in 67 candidate lysosomal related genes (LRGs), including 15 lysosomal function-related genes and 52 lysosomal storage disorder genes.
Results:
Compared with the autosomal dominant PD (ADPD) or SEOPD cohorts, a much higher proportion of patients with multiple rare damaging variants of LRGs were found in the autosomal recessive PD (ARPD) cohort. At a gene level, rare damaging variants in GBA and MAN2B1 were enriched in PD, but in SCARB2, MCOLN1, LYST, VPS16, and VPS13C were much less in patients. At an allele level, GBA p. Leu483Pro was found to increase the risk of PD. Genotype-phenotype correlation showed no significance in the clinical features among patients carrying a discrepant number of rare variants in LRGs.
Conclusion:
Our study suggests rare variants in LRGs might be more important in the pathogenicity of ARPD cases compared with ADPD or SEOPD. We further confirm rare variants in GBA are involve in PD pathogenecity and other genes associated with PD identified in this study should be supported with more evidence.
INTRODUCTION
Since SNCA mutations were first recognized as a cause of Parkinson’s disease (PD), more than 20 genes with different degrees of genetic evidence have been reported to be linked to PD. Among them, LRRK2 is the most common causative gene for autosomal dominant PD (ADPD) and late-onset PD (LOPD), and Parkin for autosomal recessive PD (ARPD) and early-onset PD (EOPD). However, LRRK2, Parkin, and other causative genes are only identified in a limited number of patients with PD. According to SNP-based polygenic risk score and the varying prevalence (0.5–2%), the heritability of the risk of developing PD was reported to be 16–36%[1]. This suggests that genetic variation from other genes that influence the pathogenesis or susceptibility of PD still needs to be identified.
Accumulating evidence has shown that heterozygous mutations in the glucocerebrosidase gene (GBA), encoding a lysosomal enzyme, are the most prevalent genetic risk factors for PD [2], suggesting an important role of lysosomal activity in the susceptibility and pathogenicity of PD [3–5]. Other lysosomal related genes, including ATP13A2 (PARK9), VPS13C (PARK23), and the newly identified prosaposin (PSAP), were suggested to be associated with PD or parkinsonism [2, 6]. In addition, rare variants in SMPD1, which causes Niemann-Pick disease type A/B, an autosomal recessive lysosomal storage disorder (LSD), were associated with PD [4, 7]. A clear association was identified between variants of several lysosomal genes, including TMEM175, CTSB, GALC, and SCARB2, and an increased risk of developing PD at a genetic level [1, 8]. Further mechanistic studies found that α-synuclein toxicity linked to lysosomal abnormalities suggested that PD was a lysosomal-related disorder [5], in which variants in hydrolase enzyme-related genes result in a loss of function is one of the major pathogenesis mechanisms [9]. Thus, rare pathogenicity/likely pathogenicity (P/LP) variants in genes linked to lysosomal function or genes causing LSDs might increase the risk or modify the phenotype for PD.
Compared with EOPD and familial PD (FPD), accumulated toxicity from aging and environmental exposure were suggested as an important cause of LOPD. Although genetic factors may play a more crucial role in EOPD and FPD than in LOPD and sporadic PD (SPD) [10], a systematic analysis of rare variants of lysosomal related genes in EOPD or FPD is not available. Furthermore, studies that identified lysosomal related genes linked to PD in Caucasians need to be replicated in other ethnic cohorts.
Based on the above, we studied rare variants of 67 candidate lysosomal related genes in a total of 1,136 participants, including 536 and 600 patients with sporadic EOPD (onset age, < 45 years) and FPD, respectively, using whole-exome sequencing (WES). We aimed to 1) reveal the proportion of rare variants of lysosomal related genes in EOPD and FPD; 2) identify the potential role of lysosomal related genes for PD at gene and allele levels; and 3) conduct a genotype-phenotype correlation analysis among patients with a different number of rare damaging variants in the candidate genes.
METHODS
Subjects
A total of 1,136 patients with PD, including 536 and 600 with sporadic EOPD (SEOPD, age of onset < 45 years) and FPD (417 ADPD and 183 ARPD), respectively, were admitted to the Department of Neurology, West China Hospital, between December 2010 and June 2019, were recruited for the current study. All patients with PD were diagnosed by experienced neurologists based on the UK Brain Bank Clinical Diagnostic Criteria for PD [11] or the 2015 Movement Disorder Society (MDS) Clinical Diagnostic Criteria [12] for PD. Patients who had at least one first-degree PD-affected relative were categorized as having familial PD. Autosomal recessive PD (ARPD) families were consanguineous, or the probands in these families had two or more affected siblings but no affected family members within two consecutive generations. In contrast, patients with at least two consecutive generations of PD in their families were classified as having autosomal dominant PD (ADPD). For the familial PD cases, only the proband were analyzed in the current study. As described in our previous study [13], the demographic and clinical data were collected. This study was approved by the ethics committee of West China Hospital, Sichuan University. All the subjects who participated in the study provided informed consent prior to participation.
DNA preparation, MLPA, and whole exome sequencing
Genomic DNA was collected from peripheral blood leukocytes via standard phenol-chloroform procedures. For multiplex ligation-dependent probe amplification (MLPA) testing for exon deletions/insertions of SNCA, Parkin, PINK1, DJ1, LRRK2, and ATP13A2, and methods of whole exome sequencing (WES) were described in our previous study [13]. Briefly, for WES, a total of 5μg DNA was fragmented to an average size of 350 bp using a Covaris LE220-plus Focused-Ultrasonicator, DNA library was constructed using a KAPA Library Amplification Kit according to the manufacture’s instruction. WES was then performed using a NovaSeq 5000/6000 S2/S1 reagent kit (Illumina) as per standard protocols.
For WES, clean data were mapped to the reference genome (GRCh37/hg19) to obtain the bam file using the BWA Picard protocol. Genotype calling was performed using Genome Analysis Toolkit’s (GATK) HaplotypeCaller software.
Sample quality control and variant quality control
The samples with a high proportion of chimeric reads (> 5%), high contamination (< 5%), poor call rates (< 90%), mean depth < 10 X, or mean genotype-quality<65 were excluded from further analysis.
For variant quality control, we restricted the data to GENCODE coding regions, where Illumina exomes surpassed 10 X mean coverage. The “PASS” variants in GATK’s variant quality score recalibration (VQSR) filter were included in further analysis. In addition, individual genotypes have to meet the following criteria: 1) genotype depth more than 10, 2) the allele balance (alternative allele cover/total allele cover) of heterozygous sites is between 0.2 and 0.8, and of homozygous sites > 0.8, 3) genotype quality (GQ) > 20. Sanger sequencing random variants that are on the lower side of quality to show that these variants are real.
Allele frequency categorization
Allele frequencies were estimated using the Genome Aggregation Database (GnomAD). Variants have a minor allele frequency (MAF) of < 0.01 or < 0.001 in GnomAD_ all and GnomAD_ EAS (East Asian) were classified as rare variants.
Candidate genes and variants annotation
Sixty-seven genes, reported in previous studies [4, 14], including 15 lysosomal function-related genes and 52 LSDs genes [15, 16], were included in this study (Supplementary Table 1). To better understand the role of lysosomal related genes in PD, we also investigated the mutation of seven established Mendelian PD genes [2], including SNCA, LRRK2, Parkin, PINK1, DJ1, VPS35, and PLA2G6 in all patients. Rare variants of all the candidate genes, which met the above criteria and were annotated as “missense”, “frameshift”, “stop-gain”, “splice acceptor” or “splice donor” were further analyzed. In accordance with a previous study [5, 17], we selected a Combined Annotation Dependent Depletion (CADD) C-score≥12.37, as rare damaging missense variants. Rare damaging missense variants and protein-truncating variants (PTVs), including “frameshift,” “splice acceptor,” “splice donor,” and “stop-gain”, were classified as potentially pathogenic variants (PPVs).
Statistical analysis
The approach of optimized sequence kernel association test (SKAT-O) implemented in R packages AssotesteR [18], were performed on an allelic basis for a genetic investigation into the collective risk of rare variants and PPVs of the 67 candidate genes for PD. Rare variants (MAF < 0.01) from 6708 controls without neurological diseases tested by WES (GnomAD_Exomes) from the east Asian cohort in the GnomAD database (version 2.1) were analyzed as the control group. At a gene level, the significant difference was corrected using the Bonferroni method (p < 0.05/67). At an allele level, the genome-wide significance p-value threshold of 5×10–8 was used.
The comparison of continuous data was assessed by the Student’s t-test. Chi-square tests were used to compare categorical variables between two groups. A two-tailed p < 0.05 was considered statistically significant. Statistical analysis was performed using SPSS version 25.0 (SPSS, Chicago, IL, USA).
RESULTS
Demographic characteristics and rare variants identified in PD
A total of 1,136 patients with PD, including 536 with SEOPD and 600 with FPD (417 ADPD and 183 ARPD), were included in this study. The demographic and clinical characteristics of the patients included in the study are presented in Table S2. Compared with patients with ADPD, a high prevalence of rigidity as the initial symptom, much slower disease progression, and much better cognition performance were seen in patients with SEOPD.
A total of 1,195 variants with a frequency below 1%and 605 variants with a frequency below 0.1%in the protein-coding regions of 67 lysosomal related genes in patients with PD were included in the analysis (Table 1 and Supplementary Table 8). A greater mean number of rare variants or rare damaging variants were found in the ARPD cohort than in the ADPD and SEOPD cohorts (Table 2). The proportion of patients carrying at least one rare variant and multiple rare variants of ARPD was also much higher than those with ADPD and SEOPD (Table 2 and Fig. 1A). Such differences tended to remain when 94 patients carrying mutations of seven PD causative genes were excluded (Supplementary Table 3 and Fig. 1B).
Candidate genes and rare variants (<1%) in Chinese sporadic early-onset PD and familial PD patients
*variants number identified in East Asian without neurological diseases from GnomAD_Exomes database; In parentheses, rare variants (< 1%) number of potentially pathogenic variants (PPVs), including PTVs and damaging missense variants.
Rare variants distribution in PD patients
B, benign; D, damaging; MAF: minor allele frequencies; PTVs: protein-truncating variants;SEOPD: sporadic early-onset Parkinson’s disease; ADPD: autosomal dominant Parkinson’s disease; ARPD: autosomal recessive Parkinson’s disease;PPVs, potentially pathogenic variants; *inparentheses, the mean variant number of PPVs, including PTVs(protein-truncating variants) and damaging missense variants (CADD≥12.37) for each patient;a patient carrying at least one rare variant; b patient carrying multiple alleles; c patient carrying at least one rare putative damaging variant; d patient carrying multiple putative damaging alleles; e comparison between ARPD and SEOPD, p = 0.018; f comparison between ARPD and ADPD,p = 0.024; g comparison between ARPD and SEOPD, p = 0.018; h comparison between ARPD and ADPD, p = 0.012.
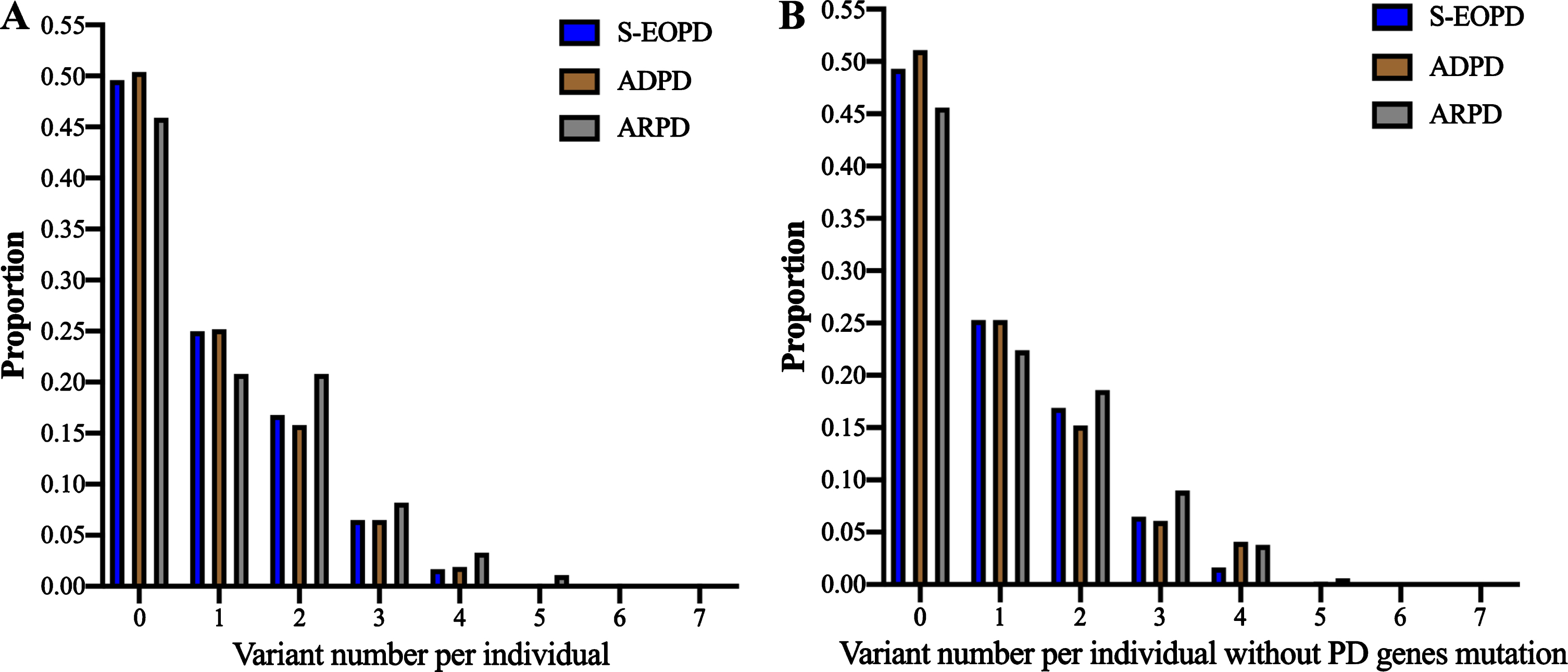
Distribution of rare damaging variants in lysosomal related genes among patients with PD (frequencies < 1%). The proportion represents the ratio of patients carrying different rare variants in each subgroup of patients with PD. Rare damaging variants, including rare PTVs and rare damaging missense variants. A) Distribution of rare damaging variants in all patients with PD; B) Distribution of rare damaging variants in patients with PD without SNCA, LRRK2, Parkin, PINK1, DJ1, VPS35, and PLA2G6 mutation.
Burden analysis
At a gene level, we performed a burden analysis for each of the 67 candidate genes for all rare variants (<1%) and the subgroup of rare PPVs (Supplementary Tables 8 and 9). We found that rare PPVs in GBA and MAN2B1 were significantly enriched in patients with PD (p = 4.1e-19; p = 0.02479, respectively); however, rare PPVs in SCARB2, MCOLN1, LYST, VPS16, and VPS13C were much less in patients (Table 3 and Supplementary Table 4). In subgroup analysis, we found rare PPVs in GBA were only significantly enriched in patients with SEOPD and ARPD, and rare PPVs in MAN2B1 were exclusively enriched in patients with ARPD. In addition, compared with controls, SCARB2 and VPS16 were much less in SEOPD and ADPD (Table 4 and Supplementary Table 5).
Genes with a Bonferroni-Corrected Significant in PD patients tested by SKAT-O
MAF, minor allele frequencies; SKAT-O, Optimized Sequence Kernel Association Test; Bonferroni, Bonferroni correction for 67 genes tested; CADD, Combined Annotation Dependent Depletion, * including PTVs (protein-truncating variants) and damaging missense variants (CADD≥12.37); #Controls from GnomAD_Exomes database in East Asian population.
Genes with a Bonferroni-Corrected Significant in sporadic early-onset PD, ADPD and ARPD tested by SKAT-O
CADD, Combined Annotation Dependent Depletion; MAF: minor allele frequencies; *p-value conducted by Optimized Sequence Kernel Association Test (SKAT-O); #including PTVs (protein-truncating variants) and damaging missense variants (CADD≥12.37); SEOPD, sporadic early-onset Parkinson’s disease, alleles = 1,072; ADPD, autosomal dominant Parkinson’s disease, alleles = 834; ARPD, autosomal recessive Parkinson’s disease, alleles = 366; Controls from GnomAD_Exomes database in East Asian population, alleles = 13,416; Bonferroni, Bonferroni correction for 67 genes tested; Bold means significant.
At an allele level, the rare damaging variant, p. Leu483Pro (rs421016) in GBA, was found to have genome-wide significance (p < 5e-8, OR = 11.1, 95%CI: 6.0–20.3). Three other PPVs in GBA, p.Asp448His (rs1064651), p.Asn227Lys (rs381418), and p.Gly241Arg (rs409652), were much more common in patients with PD. However, they did not pass to genome-wide significance (Supplementary Table 6).
Genotype-phenotype correlation analysis
In total, 560, 277, 194, 77, 23, four, and one patients carried zero, one, two, three, four, five, and six rare PPVs, respectively. Their clinical features are shown in Table 5. No significant differences in the sex distribution, age of onset, initial symptom, Montreal Cognitive Assessment (MoCA), non-motor symptoms scale (NMSS), Parkinson’s Disease Questionnaire-39 (PDQ39), frontal assessment battery (FAB), Hamilton Depression Rating Scale (HAMD), and Hamilton Anxiety Rating Scale (HAMA) scores were found among patients carrying different numbers of rare PPVs. Considering the effect of PD gene mutations, we grouped the patients with PD by those with and without gene mutations. We also did not find any differences in the phenotypes between patients with and without lysosomal related gene variants whether they carried PD causative gene mutations or not (Supplementary Tables 7 and 10).
Genotype-phenotype correlations among patients with none, only one variant and multiple variants
T, tremor; R, rigidity; O, bradykinesia, gait disturbance and non-motor symptoms; UPDRS, Unified Parkinson Disease Rating Scale; MoCA, Montreal Cognitive Assessment; NMSS, non-motor symptoms scale; PDQ-39, Parkinson’s Disease Questionnaire-39; FAB, frontal assessment battery; HAMD, Hamilton Depression Rating Scale; HAMA, Hamilton Anxiety Rating Scale; In parentheses, the number of patients whose data was available.
DISCUSSION
In this study, we comprehensively analyzed rare variants in a subset of 67 lysosomal functional-related and LSDs genes in patients with PD, including SEOPD and FPD. We found that patients with ARPD might carry more rare variants of lysosomal relative genes compared to patients with ADPD or SEOPD. We further confirmed that GBA is the most important lysosomal related gene that increases the risk for PD, although rare PPVs in MAN2B1 might involve in ARPD. Our results suggest that gene or allele dosage effects of lysosomal related genes do not play a critical role in the phenotype of patients with PD based on a comprehensive analysis of a large Chinese population.
As described above, dysfunction of lysosomes is involved in the pathogenicity of PD, which was supported by the evidence from genetic, biochemistry, and mechanism studies [9]. However, at a genetic level, the main evidence of the association between PD and lysosomes comes from genome-wide association studies, which implicates more common risk alleles in LSD genes that contribute to PD, such as SCARB2, GALC [20], VCP13C [21] At a rare variant level, only two studies from Caucasians systematically reported the excessive burden of several lysosomal related genes in PD [4, 14]. However, except for GBA, other identified lysosomal related loci suggested to be susceptible to PD have had conflicting findings. The identified loci, SMPD1 and CTSD from the International PD Genomics Consortium (IPDGC) [4], were not replicated in an ethnically homogeneous sample of people from Germany [14]. On the other hand, as reported in our previous studies, the association between polymorphisms of SCARB2 [22] and GALC [23] and PD identified in Caucasians was not replicated in the Chinese population. Thus, further studies that investigate the role of the susceptibility loci, or the connection between lysosomal related genes and PD from different ethnic origins, are urgently needed.
In the current study, the 67 candidate genes included 15 genes with a function linked to lysosomal degradation reported in the Germany study [14], and 52 genes causing LSDs reported in IPDGC [4], therefore our study more comprehensively represents the relationship between lysosomal function and PD. Strict Bonferroni correction was performed to limit the likelihood of type I errors. Furthermore, in the German study [14], only patients with sporadic PD with an age of onset older than 45 years were included. In contrast, in the current study, patients with SEOPD and FPD were included. These patients are more likely to have a potential genetic contribution, as reported in the IPDGC study [4]. We also analyzed the characteristics of subgroups of patients with PD that carried different numbers of rare variants in lysosomal related genes according to different inheritance pattern. We conducted a genotype-phenotype correlation analysis in patients with different numbers of rare variants considering the gene or allele dosage effect. Moreover, we considered the effect of PD gene mutations on the phenotype in patients with and without rare variants.
Interestingly, a much higher mean number of rare variants in each patient with ARPD was found, compared with ADPD or SEOPD. Although the IPDGC study [4] investigated the characteristics of the rare variant distribution of LSD genes in the younger-onset and FPD in the discovery stage, they did not analyze the rare variant distribution in these subgroups of PD according to different inheritance pattern or whether they have familial history. Our study found that the average variant burden among ARPD cases was more than 0.2 higher than that in ADPD or SEOPD, indicating that the cumulative dysfunction of lysosomal proteins by multiple alleles contributed to the risk of ARPD. This was further supported by the finding that the proportion of patients with more than one rare variant in ARPD was much higher than that in ADPD or SEOPD. This finding was also found when patients with PD causative gene mutations were excluded, which supports the polygenic effect of loss of function, such as lysosomal dysfunction, might be responsible for some PD cases, particularly those with ARPD.
We did not investigate the total burden of all candidate lysosomal related genes because single gene is a better functional unit in the pathogenesis of PD than gene sets, in consideration of diverse functions or bidirectional effects of different genes, or negative findings caused by the effect of dilution from several unrelated genes, as described in the German study [14]. As shown in our study, the frequencies of rare variants in some genes, such as SCARB2, MCOLN1, LYST, VPS16 and VPS13C, were much higher in the controls than in the patients with PD. This was partially supported by the IPDGC study as they did not find any differences in the distributions of non-synonymous missense variants, damaging missense variants, or loss of function variants between PD and controls when the frequency of rare variants was less than 1%[4]. In the individual gene analysis, we found that rare variants (<1%) in GBA increased the risk for PD, even in the ultra-rare variants (<0.1%, p. L483P in GBA was excluded), which was consistent with other studies in Caucasian [4, 14] and Asian populations [13]. Pathologically, the loss of function of GBA leads to alteration of α-synuclein metabolism, a hallmark of PD [24]. Interesting, rare PPVs in MAN2B1 were significantly enriched in ARPD. Previous study found the majority of missense variants caused the deficiency of the enzyme lysosomal alpha-mannosidase, supported its pathogenicity in alpha-mannosidosis [25], but whether a higher rate of progressively developing parkinsonian features in relatives of patients or patients with alpha-mannosidosis, an autosomal recessive lysosomal storage disorder, need more evidence. In addition, our burden analysis did not support the excessive burden of VPS13C variants in Chinese patients, but more higher proportion of rare variants in controls, which might be explained in part by the observation that rare damaging variants in VPS13C contribute to the development of the recessive form of PD [26]. However, findings that rare variants in LAMP1, TMEM175, CTSD, SLC17A5, and ASAH1, which were reported in Caucasian [4, 14] to increase the risk for PD, were not replicated in Chinese population. The differences in ethnic origin, sample size of the recruited patients, and the phenotype of PD might have resulted in these differences. Considering the function of lysosomes, the activity of hydrolase is critical in the process of degradation, so rare PTVs causing a loss of function were analyzed in our study (data not shown). However, only PTVs in SMPD1 were much more in PD (p = 0.0038), which was partially consistent with the findings from the IPDGC study [4] and another Asian cohort study [7]. Although some variants in SMPD1, such as p.L302P and p.R496L, were strongly associated with a highly increased risk for PD in Caucasian [27, 28], the distribution of rare PPVs including PTVs and rare damaging missense variants in SMPD1 were not different between PD cases and controls in our study, indicating that the effect of SMPD1 in PD was varying among different ethnic origin. In addition, incomplete penetrance might be another reason for the same proportion of rare variants identified in controls.
We first analyzed the genotype-phenotype correlation in patients with PD that had a different number of rare PPVs in lysosomal related genes. Previous studies found that GBA variants may modify the clinical manifestations of PD, including age of onset [29], cognition [30], or disease progression [31], and the gene dosage effect was also suggested in patients with multiple alleles of GBA in our previous study [32]. However, no significant differences in age of onset, initial symptom, cognition, and non-motor symptoms between patients with and without rare variants were found, whether they carried PD causative gene mutation or not. In addition, gene dosage effects were not identified when patients carrying different numbers of rare PPVs were analyzed. The above finding might be explained by the fact that some lysosomal genes were not related to the risk for PD, as per the findings in the rare variant burden analysis. Therefore, an analysis of the genotype-phenotype correlation should be conducted in single or specific gene/genes in the future, when the sample size with rare variants of candidate genes is sufficient.
Although we conducted a comprehensive genetic screening for rare variants in 67 lysosomal related genes and genotype-phenotype analysis in a total of 1,136 patients with SEOPD and FPD, these results should be interpreted with caution. Firstly, in our burden analysis, the consensus deleteriousness score was used to help exclude rare but benign missense variants in PD and the controls from the database. However, the pathogenicity of those predicted to be a deleterious variant should be further confirmed based on more experimental evidence. Secondly, using data from the East Asian GnomAD_Exomes database rather than ethnically age- and sex-matched controls may also contribute to these negative findings. Thirdly, the methods of variant detection among the patients and controls, the definitions of SEOPD and FPD, also caused the false-negative or false-positive results between patients and controls. Lastly, etiological heterogeneity, incomplete penetrance, and late-onset diseases make it difficult to detect associations between rare variants and the disease in case-control studies. Overall, we do not deny lysosomal dysfunction is involved in PD.
CONCLUSION
In conclusion, rare variants in lysosomal related genes might be more important in the pathogenicity of ARPD cases compared with ADPD or SEOPD. The rare variant burden analysis further confirmed that GBA, the most important lysosomal related gene, increased the risk for PD. Gene dosage effects from all the candidate lysosomal related genes were not found to play a critical role in the phenotype of patients with PD.
Footnotes
ACKNOWLEDGMENTS
The authors appreciate all cohort individuals and their families for their participation in this study. We thank Ya-Xin Chen, Department of Respiratory and Critical Care Medicine, West China Hospital, Sichuan University, Chengdu, Sichuan Province, China, for providing the methods of the optimized sequence kernel association test (SKAT-O).
This study was supported the National Key Research and Development Program of China (grant no. 2016YFC0901504 and 2018YFC1312001), and the 1.3.5 project for disciplines of excellence, West China Hospital, Sichuan University (Grant No. ZYJC18038 and Grant No. ZYJC18003).
CONFLICT OF INTEREST
The authors report no conflicts of interest.