
Abstract
Background:
Melanopsin-expressing intrinsically photosensitive retinal ganglion cells (ipRGCs) signal the environmental light to mediate circadian photoentrainment and sleep-wake cycles. There is high prevalence of circadian and sleep disruption in people with Parkinson’s disease, however the underlying mechanisms of these symptoms are not clear.
Objective:
Based on recent evidence of anatomical and functional loss of melanopsin ganglion cells in Parkinson’s disease, we evaluate the link between melanopsin function, circadian, and sleep behavior.
Methods:
The pupil light reflex and melanopsin-mediated post-illumination pupil response were measured using chromatic pupillometry in 30 optimally medicated people with Parkinson’s disease and 29 age-matched healthy controls. Circadian health was determined using dim light melatonin onset, sleep questionnaires, and actigraphy. Ophthalmic examination quantified eye health and optical coherence tomography measured retinal thickness.
Results:
The melanopsin-mediated post-illumination pupil response amplitudes were significantly reduced in Parkinson’s disease (p < 0.0001) and correlated with poor sleep quality (r2 = 33; p < 0.001) and nerve fiber layer thinning (r2 = 0.40; p < 0.001). People with Parkinson’s disease had significantly poorer sleep quality with higher subjective sleep scores (p < 0.05) and earlier melatonin onset (p = 0.01). Pupil light (outer retinal) response metrics, daily light exposure and outer retinal thickness were similar between the groups (p > 0.05).
Conclusion:
Our evidence-based data identify a mechanism through which inner retinal ipRGC dysfunction contributes to sleep disruption in Parkinson’s disease in the presence of normal outer retinal (rod-cone photoreceptor) function. Our findings provide a rationale for designing new treatment approaches in Parkinson’s disease through melanopsin photoreceptor-targeted light therapies for improving sleep-wake cycles.
Keywords
INTRODUCTION
Non-motor symptoms including circadian and sleep disturbances are predominant in people with Parkinson’s disease (PD) [1], and can predate and exaggerate motor dysfunction [2]. Sleep related problems often manifest as poor sleep quality, excessive daytime sleepiness [3, 4], obstructive sleep apnea and REM sleep behavior disorder [2]. In addition, people with PD experience abnormalities in their melatonin secretion pattern [5]. The pathophysiology of circadian and sleep disruption in PD are however, not clearly understood.
Melanopsin expressing intrinsically photosensitive Retinal Ganglion Cells (ipRGCs) provide major input to circadian and sleep centers [6]. IpRGCs combine inputs from rod and cone photoreceptors to mediate the human pupil light reflex [6, 7] and signal to the suprachiasmatic nucleus (SCN) via the retino-hypothalamic tract for photoentrainment [8]. IpRGCs have direct pathways to the ventrolateral preoptic area (VLPO) for sleep induction [9]. We provided the initial evidence of ipRGC dysfunction in people with PD [10]. Moreover, anatomical and histological examination of post-mortem PD eyes reveal morphological changes in ipRGCs [11], retinal thinning [12] and deposition of phosphorylated α-synuclein in the inner retina [13]. We therefore hypothesize that functional and structural changes in ipRGCs provide a retinal origin for circadian and sleep disruption in PD.
Here we apply chromatic pupillometry to quantify ipRGC function and use objective and subjective measures of circadian and sleep health in a cross-sectional study of people with different PD severities in comparison to healthy, age-matched controls.
METHODS
Participants
Thirty-three people with PD (age range: 52 – 75 years; 67.3±8 years; mean±SD) were recruited from the Parkinson’s study group at Queensland University of Technology (QUT) and the Parkinson’s Queensland support group. Individuals diagnosed by their treating neurologist were optimally medicated (Levodopa Equivalent Daily Dosage = 482.92±273.9; mean±SD). People with PD had a disease duration between 8 months– 25 years (median = 7.2 years) and were grouped into mild (Stage I and II; n = 10), moderate (Stage III; n = 10) and severe (Stage IV; n = 10) disease stages according to the Movement Disorders Society Unified Parkinson’s Disease Rating Scale (MDS - UPRDS) and the Hoehn and Yahr scale[14, 15]. Cognition was assessed using the Addenbrooke’s Cognitive examination (ACE, Version IV, and Revised 2004).
Twenty-nine age-matched healthy controls (range: 41– 73 years; 63±9 years; mean±SD) with no history of ocular disease, no uncontrolled systemic disease, sleep or mood disorders were recruited from the QUT health clinics, staff and students. In both test cohorts, we excluded people who were shift workers or travelled across two or more time-zones in the last 90 days, with diabetes, sleep apnea, restless leg syndrome, on antidepressants and sleep/wake promoting medication, cataracts > grade 2, multifocal or blue blocking intraocular implants that can potentially affect the outcomes of the pupil response, circadian health and sleep [16].
Participants completed an ophthalmic examination with optical coherence tomography (RS 3000-Advance, HD OCT, Nidek, USA) and sleep questionnaires (PSQI and ESS) on their first visit. Participants received a sleep log to assess daily sleep wake time, an actiwatch (GENE Active, Activinsights, UK) to measure daily light exposure and activity and a Saliva collection kit (Salivettes, Sarstedt, AG&Co) for dim light melatonin onset (DLMO) determination. Chromatic pupillometry was performed on the second visit.
Experimental protocols were conducted in accordance with the tenets of the declaration of Helsinki and received approval from the QUT Human Research Ethics Committee (#1700000699). The research purpose and study protocols were explained in detail and written informed consent was obtained from all participants.
Pupillometry
A Maxwellian-view pupillometer was used to measure the pupil light response as established in our laboratory [17, 18]. Participants were dark adapted (10 min) and pupil responses measured in the dark (<1 lux). In accordance with our standard protocols [17–19], pupil recording included a 10 s pre-stimulus (baseline pupil diameter) followed by a 1 s light pulse (pupil light response, PLR) and 40 s after light offset (post-illumination pupil response, PIPR). Narrowband 50° diameter foveated stimuli included blue and green lights with high melanopsin excitation (λmax = 460 nm blue = 7998 α-opic lux, retinal irradiance = 14.98 log.quanta.cm-2.s-1;λmax = 519 nm green = 6985.78 α-opic lux, retinal irradiance = 15.21 log.quanta.cm-2.s-1) and red lights with low melanopsin excitation (λmax = 630 nm red = 4.05 α-opic lux, retinal irradiance = 15.34 log.quanta.cm-2.s-1) having a corneal irradiance of 15.5 log.quanta.cm-2.s-1. Pupil measurements were repeated twice for all wavelengths (red, green and blue) with a 2 min interval between stimuli such that the PIPR of the previous recording does not affect the pupil response of successive recordings [17]. Pupil recordings were performed between 10 am and 5 pm to minimize the effect of circadian variation [18].
Blink and lid artefacts in the raw pupil data were manually extracted and linearly interpolated using customized software (MATLAB R2016b, V9.1.0, MathWorks, USA). Data were normalized to baseline pupil diameter to control for pupil size variations between individuals [17, 20]. The PLR during light onset was quantified using the transient PLR and peak pupil constriction amplitude metrics. The melanopsin-mediated 6s PIPR after light offset was quantified in accordance with established protocols [17] and international standards in pupillography [19].
Circadian rhythm assessments and sleep
Dim light melatonin onset
Salivary melatonin was used to determine circadian phase and completed at home under dim light illumination (<10 lux) [21]. We used a sparse sampling protocol to estimate circadian phase [22]. Participants received seven labelled Salivettes and instructions how to collect their saliva. Samples were collected every hour starting 6 h before habitual sleep onset and concluded 1 h after habitual sleep onset in accordance with established protocols. Melatonin concentrations were determined by radioimmunoassay (Bühlmann Laboratories AG, Switzerland) [23]. The lowest limit of quantification (LLoQ) was 4.3 pico-molar (pM). To estimate circadian phase, the melatonin data for each participant as a function of time was described using a skewed baseline cosine function (SBCF) model (Equation 1) [22], where
and t = time (radians), b = baseline salivary melatonin, H = the amplitude above baseline, c = width, Φ= phase (radians) and v = skewness. The resulting curve was used to determine DLMO which was defined when the melatonin concentrations reached 0.01pM above the salivary baseline [18]. The difference between the melatonin onset and habitual sleep onset (derived from sleep diary) was defined as the phase angle of entrainment.
Actigraphy
A wrist-worn actiwatch (GENE Active, Activinsights, UK) captured the participant’s light exposure and physical activity (but not sleep parameters) over 14 days. Data were extracted using GENE Active software (GENE Active V3.1, Activinsights Ltd, UK). Raw data were screened to remove artefacts; such as daytime measurements showing complete darkness (0 lux) or evidence of data generated during non-wear times. The average day and night light exposure patterns, the time to first and last light, and global solar exposure were calculated using data obtained from WillyWeather and the Australian Government Bureau of Meteorology. To quantify average light exposure and activity, we used a 60 s sampling rate [24]. Activity levels were recorded and categorized as sedentary, light, moderate and vigorous activity using the Activinsights software.
Sleep assessment
The Pittsburgh Sleep Quality Index (PSQI), Epworth Sleepiness Scale (ESS) questionnaires and a sleep diary determined sleep behavior. In the PSQI, the total global score was calculated [25]. A two-factor model was used to report “perceived sleep quality” and “perceived sleep efficiency” [26]. Daytime sleepiness was measured using the ESS. In addition, participants documented in their sleep diary, the time to bed, time taken to fall asleep, number of minutes awake at night, first wake time, final awakening, afternoon naps and non-wear times of the actigraph. Sleep efficiency (actual sleep time/Total time in bed*100) and an individual’s chronotype were determined using daily sleep and wake times. While pupillary unrest and the pupillary unrest index (PUI) are an objective measure of sleepiness [27, 28], our previous study [10] did not detect a significant difference in patients with PD compared to healthy controls during the day and so the PUI was therefore not measured in this study.
Statistical analysis
Statistical analyses were performed using SPSS (IBM SPSS, version 23; IBM Corporation, USA) and GraphPad Prism (GraphPad Software, Inc., USA). Data were screened for normality using the Shapiro-Wilk test. Means of the parametric data between the four groups (control, mild, moderate and severe PD) were compared using one-way ANOVA (post-hoc: Tukey’s multiple comparisons) and non-parametric data using Kruskal-Wallis (Dunn’s multiple comparisons). Post-hoc analyses were performed to compare the between the group means when significant effects occurred. Independent t-test (parametric data) and Mann-Whitney U test (non-parametric data) were performed to compare the means between PD and healthy controls. The relationship between the PIPR amplitude, sleep quality or mean RNFL thickness was evaluated with linear regression.
RESULTS
Of the 33 people with PD, three were excluded on their first visit; one each due to a suspicious optic disc, restless leg syndrome and use of melatonin supplements; no controls were excluded during the eligibility assessment. Hence, the study sample included 30 people with PD (mean±SD; 63.7±8.3 years, 16 men) and 29 age-matched healthy controls (mean±SD: 63±9 years; 11 men). Both, PD and control groups did not differ in visual acuity (≥6/7.5 in both groups). The clinical characteristics and ophthalmic parameters are given in Table 1. The mean retinal nerve fibre thickness was significantly reduced in all groups of participants with PD compared to controls (F3,49 = 14.5; p < 0.001; post-hoc Tukey’s HSD for controls versus mild p = 0.01, moderate; p < 0.001 and severe; p < 0.001), but not significantly different within moderate, mild and severe stages of PD groups (Tukey’s HSD; p > 0.05). The outer retinal layer thickness (F3,49 = 2.34; p = 0.67) and total retinal thickness at 3 mm eccentricity (F3,49 = 1.72; p = 0.17) did not differ between groups.
Clinical characteristics and ophthalmic parameters of healthy controls and participants with mild, moderate and severe Parkinson’s disease
SD, standard deviation; RNFL, retinal nerve fibre layer thickness; Asterisks (*) denote statistical significance (p < 0.05) between healthy controls and overall Parkinson’s disease group (p < 0.05). The symbol §denotes the statistical difference (p < 0.05) between healthy controls versus individual PD subgroup (post-hoc pairwise comparisons).
Pupil light reflex
Table 2 gives the descriptive statistics (mean ± 95% confidence limits) of the pupil light responses for PD subgroups and healthy controls. Figure 1 shows the averaged pupil response (±95% confidence limits) of the PD subgroups compared to the mean pupil responses of the control participants (±95% confidence limits) for the blue (460 nm) stimulus with high melanopsin excitation. Figure 2 (a-i) outlines the individual data for the PLR and PIPR group comparisons in box plots. There were no significant differences between the PLR and PIPR metrics within PD subgroups. In people with PD, the transient PLR (red stimulus: F3,52 = 1.04, p = 0.13; blue: F3,54 = 2.26, p = 0.09; green: F3,54 = 1.20, p = 0.31; Tukey’s HSD) and the peak pupil constriction amplitudes for all wavelengths (red: F3,55 = 1.04, p = 0.38; blue: F3,52 = 1.74, p = 0.16; green: F3,55 = 2.58, p = 0.07; Tukey’s HSD) (Fig. 2, a-f) were normal, indicating normal outer retinal function in PD. However, the intrinsic ipRGC response (Fig. 2 h; 6sPIPR metric) to the blue (F3,53 = 20, p < 0.0001; Tukey’s HSD) and green stimuli (F3,51 = 11.93; p < 0.0001; Tukey’s HSD) (Fig. 2i) with high melanopsin excitation were significantly reduced in all participants with PD compared to controls.
PLR and PIPR metrics for 630 nm (red), 460 nm (blue) and 519 nm (green) stimuli in participants with mild, moderate, severe PD and healthy controls
95% CL, 95% confidence limits; PLR, pupil light response; PIPR, post-illumination pupil response; Asterisks (*) denote statistical significance (p < 0.05) between healthy controls and overall Parkinson’s disease group (p < 0.05). The symbol (§) denotes the statistical difference (p < 0.05) between healthy controls versus individual PD subgroup (post-hoc pairwise comparisons).
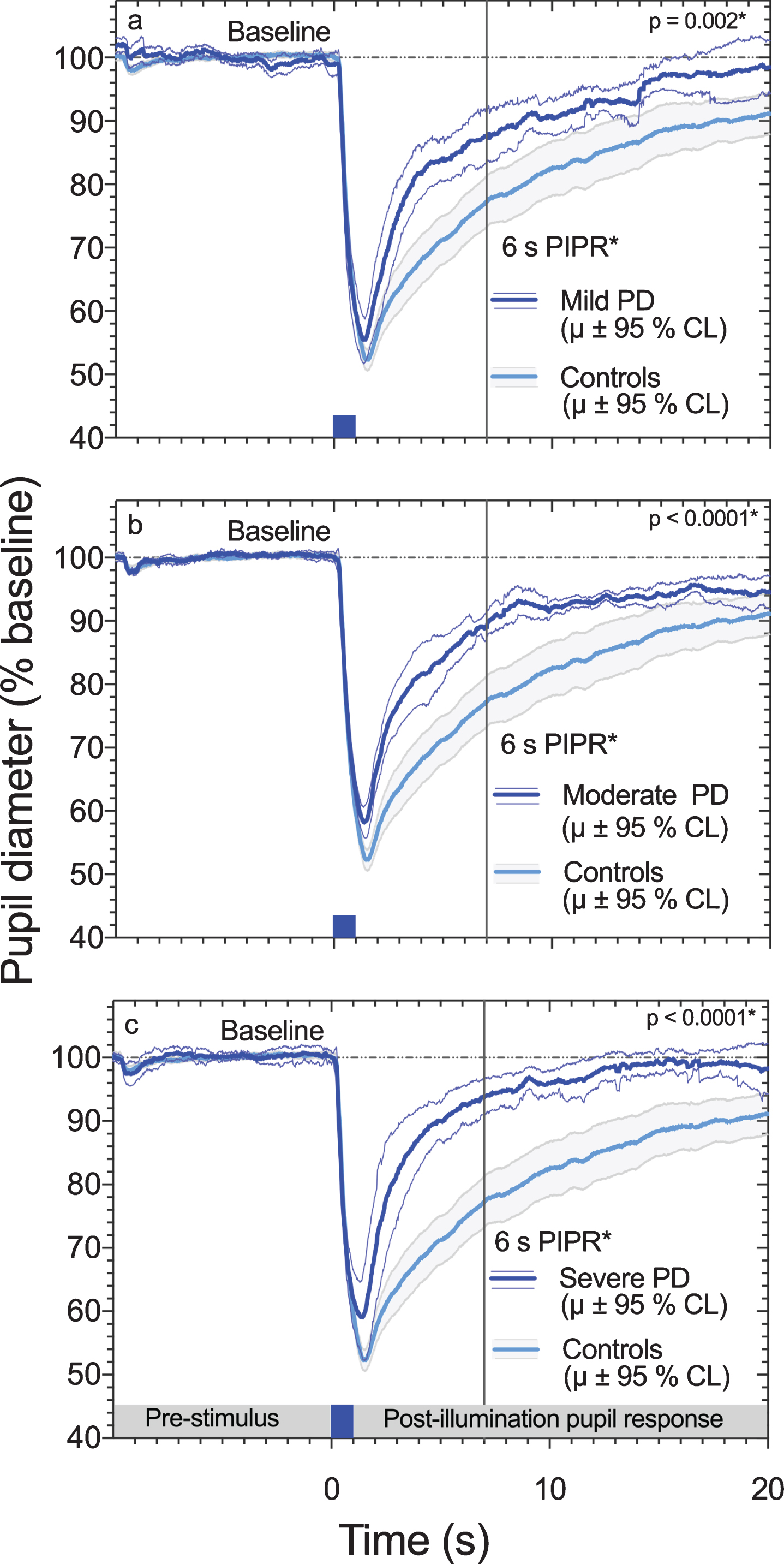
Average pupil light reflex (± 95% CI) for controls (n = 29), mild PD (n = 10); panel A, moderate PD (n = 10); panel B and severe PD (n = 10); panel C in response to a 1 s short wavelength (460 nm), 15.5 log quanta.cm-2.s-1 stimulus (blue bar at 0 s) presented in a 50 degree diameter field. The melanopsin mediated 6 s PIPR amplitudes (vertical grey line) were significantly lower (smaller % to pupil baseline diameter) in people with PD compared to controls.
Dim light melatonin onset
Actigraphy confirmed DLMO was collected in dim light (PD = 5.36±3.02 lux, controls = 3.5±4.0 lux; mean±SD; p = 0.52). Salivary samples of 30 people with PD (210 samples) and 24 healthy controls (168 samples) were assayed to determine the melatonin onset. The sensitivity of the assay was 4.3 pM. Saliva samples were assayed in duplicate. The intra-assay coefficient of variation of the assays was determined to be 8.4%. The inter-assay coefficient of variation of the low concentration quality control was 1.6%, and the inter-assay coefficient of variation of the high concentration quality control was 5.2%. Twelve people with PD (9 participants with low secretor status had melatonin concentrations<detection level, 3 with irregular melatonin concentrations) and nine controls with low secretor status were excluded thereby resulting in 126 samples (PD) and 105 samples (controls) for DLMO analysis.
There were no significant differences between PD subgroups for the phase angle of entrainment (F2,18 = 0.06; p = 0.93; Tukey’s HSD) or DLMO (F2,15 = 1.64; p = 0.22; Tukey’s HSD) and PD subgroups were pooled for further analysis. In the PD group, the phase angle was significantly longer by 58 min (Mann-Whitney U: PD = 2 h 12 min±1 h 19 min; controls = 1 h 15 min±42 min; mean±SD; p = 0.03) and the DLMO was significantly earlier by 1 h 5 min (Mann-Whitney U test: PD clock time = 19.50 pm; controls = 20.55 pm median; p = 0.01) compared to the controls (Table 3, Fig. 3). The habitual sleep onset time of the PD group (clock time 21.56 pm±40 min; mean±SD) was similar to controls (clock time 22.20 pm±24 min; mean±SD). The midpoint of sleep derived from the sleep diary indicated that both groups had similar chronotypes (morning type).
Dim light melatonin onset and Sleep profile in healthy controls and people with Parkinson’s disease
SD, standard deviation; PSQI, Pittsburgh Sleep Quality Index; ESS, Epworth Sleep Scale; Asterisks (*) denote statistical significance (p < 0.05) between healthy controls and overall Parkinson’s disease group (p < 0.05). The symbol (§) denotes the statistical difference (p < 0.05) between healthy controls versus individual PD subgroup (post-hoc pairwise comparisons).
Actigraphy
The daily average day light exposure (first to last light) was not significantly different between the groups (F3,49 = 0.15; p = 0.92; PD: 1178±366 and controls: 1239±491; mean Lux±SD). Mean night light exposure did not vary between groups (F3,49 = 0.29; p = 0.82; PD: 28±18; controls: 31±24; mean Lux±SD). The global solar exposure (GSE) and daily time spent in bright outdoor light (>1000 Lux) did not differ between or within groups (p > 0.05).
There were no differences in the moderate, light or vigorous activities in the participants with mild, moderate and severe PD compared to controls. There was, however, a significant increase in the time of sedentary activity in participants with severe PD compared to controls (Dunn’s MC: PD: 624 min; controls: 536 min median; p = 0.02) but not significantly different from mild and moderate PD.
Sleep assessment
Analysis of mean PSQI global scores identified a significantly higher number of poor sleepers in the PD group (70%) compared to controls (12%) (Kruskal-Wallis: df = 3, p < 0.001, Dunn’s MC). The PSQI sleep factor components demonstrated on average poorer sleep quality (F3,55 = 17.55; p < 0.0001; Tukey’s HSD) and reduced sleep efficiency (Kruskal-Wallis: df = 3; p < 0.001, Dunn’s MC). The PSQI scores and factor score analysis did not differ within PD subgroups (Dunn’s MC: p > 0.05) (Table 3). ESS scores were significantly higher in the moderate and severe PD but not in mild PD compared to controls (F3,55 = 3.25; p = 0.02; Tukey’s HSD). Within PD subgroups, the ESS scores were significantly higher for participants with severe stages of PD compared to participants with mild PD (Tukey’s HSD, p = 0.03). There were no differences in the ESS scores between participants with mild and moderate PD (Tukey’s HSD: p = 0.15) (Table 3).
The sleep diary revealed reduced sleep efficiency in all subgroups of PD relative to controls (F3,51 = 6.19; p = 0.01; Tukey’s HSD), but no differences between the PD subgroups (Tukey’s HSD; p > 0.05) (Table 3). The increase in number of minutes awake at midnight was evident only in moderate and severe PD, with no differences between control participants and those with mild PD (Kruskal-Wallis: df = 3, p = 0.01; Dunn’s MC, controls versus mild PD: p > 0.05; moderate and severe PD: p < 0.05). Sleep duration did not differ in participants with mild and moderate PD compared to controls. However, it was significantly reduced by one hour in participants with severe PD (Kruskal-Wallis: df = 3; Dunn’s MC, controls versus mild and moderate; p > 0.05 and severe PD; p = 0.02). Sleep onset and wake time were similar between groups, however, people with PD experienced increased sleep fragmentation after sleep initiation. There were no statistical differences in daytime napping between controls and PD (KW test; df = 3; p = 0.28). We observed that 30% of people with mild PD reported nap times between 15– 30 min, 20% of people with moderate PD reported daytime napping between 10– 30 min and 40% of people with severe PD reported between 15– 35 min. There were no correlations between daytime naps and DLMO (mild PD: r = – 0.56; p = 0.32; moderate: r = – 0.02; p = 0.96; severe: r = – 0.20; p = 0.74; Spearman’s correlation) or phase angle of entrainment (mild: r = – 0.10; p = 0.87; moderate: r = – 0.02; p = 0.96; severe: r = 0.35; p = 0.55; Spearman’s correlation).
Ophthalmic parameters and sleep quality
Linear regression determined that lower PIPR amplitudes were associated with poorer sleep quality (r2 = 0.33; p < 0.001). In addition, the decline in the PIPR amplitude for the blue stimulus correlated with the decreasing RNFL thickness (r2 = 0.40; p < 0.001) indicating that impaired melanopsin-mediated ipRGC function is associated with a thinner RNFL. There was no significant relationship between the pupil parameters and DLMO, phase angle of entrainment, total PSQI score, sleep efficiency or ESS.
DISCUSSION
This study provides evidence of a retinal contribution to disrupted sleep behavior in people with Parkinson’s disease. The ipRGC-mediated PIPR amplitude is reduced (Figs. 1 and 2) and associated with poor sleep quality (PSQI) in optimally medicated people with PD compared to healthy control participants. IpRGC dysfunction increases with retinal nerve fiber layer thinning in PD. While environmental light exposure is similar in PD and controls, people with PD show an earlier dim light melatonin onset (DLMO) (Fig. 3) that we infer may be due to abnormal transmission of the ambient light signal via ipRGCs.
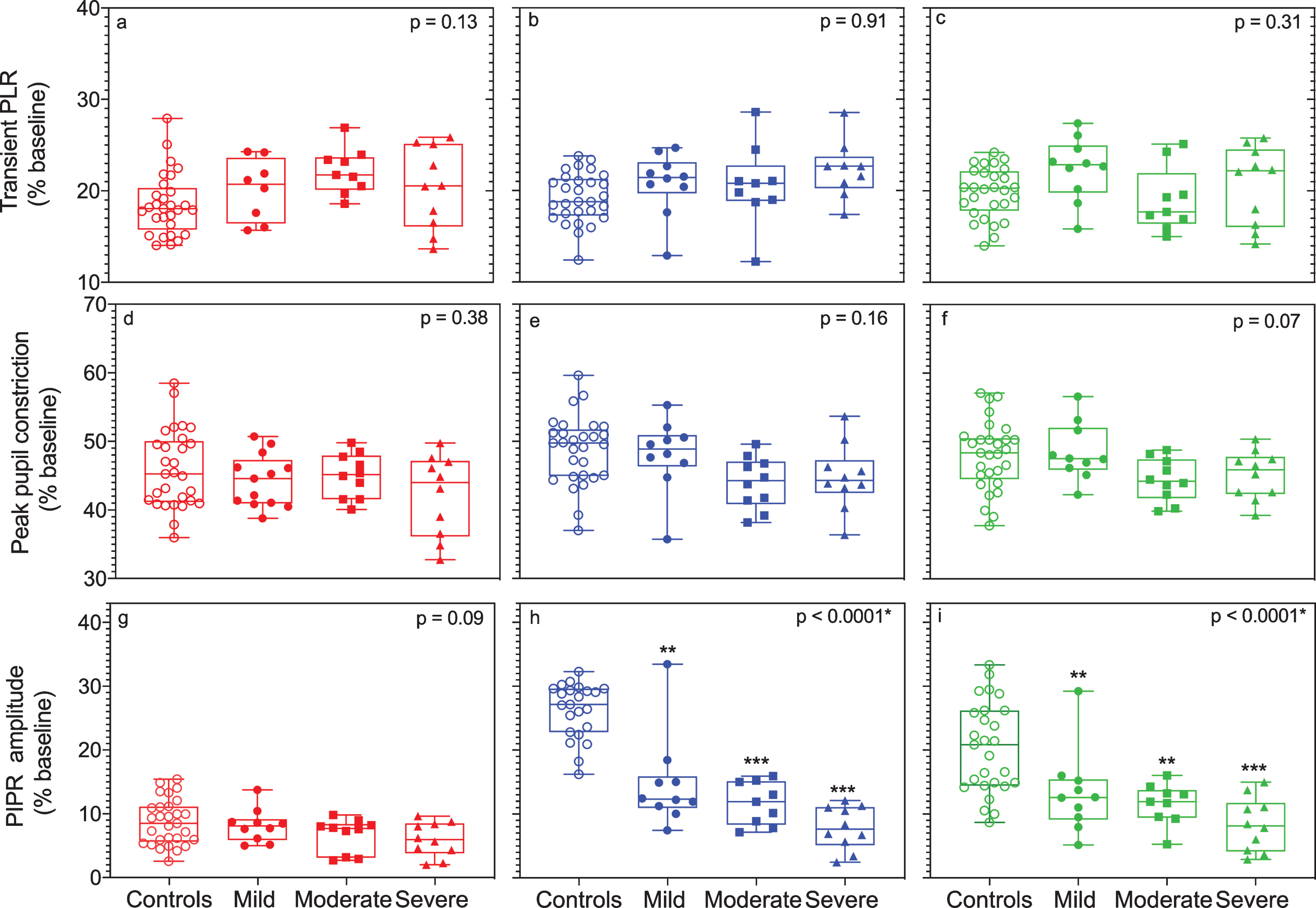
Box and whisker plots show the median interquartile and range of the transient PLR, peak pupil constriction and PIPR amplitudes for red (630 nm, left column), blue (460 nm, middle column), and green (519 nm, right column) wavelength stimuli for participants with mild, moderate and severe PD in comparison to controls. The PLR and PIPR metrics are represented as a percentage (%) of the baseline pupil diameter. There was no significant difference between groups for the transient PLR or peak pupil constriction with red, blue and green stimuli (panels a– f). The PIPR amplitudes for blue (panel h) and green stimuli (panel i) were significantly reduced in the PD groups compared to the control group (*p < 0.05, **p < 0.01, ***p < 0.001).
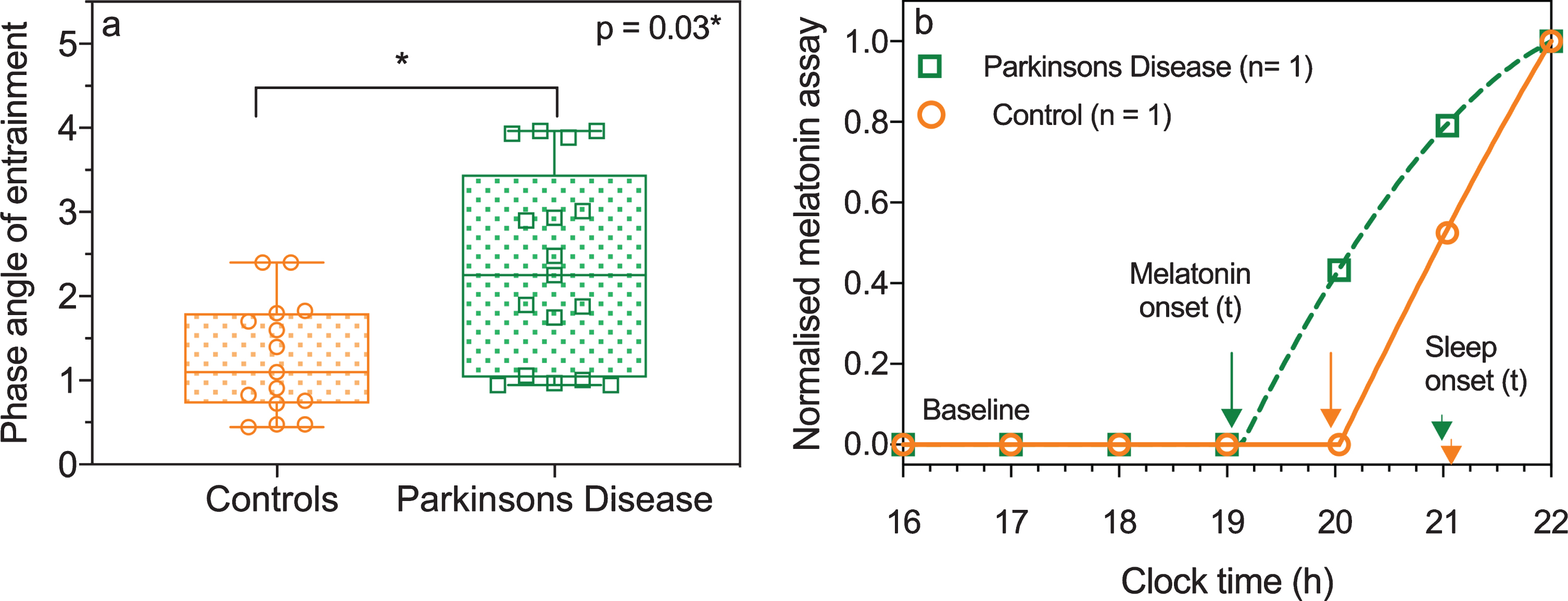
A) Box and whisker plots show the median, interquartile and range of phase angle of entrainment in controls (circular symbols) and people with Parkinson’s disease (square symbols). The entrainment phase angle was delayed indicating circadian phase advance in people with PD compared to the control group. B) A representative example of the SBCF model for an individual with PD (squares) and a healthy control (circles) who had similar sleep onset. The arrows indicate the melatonin onset times, which are at 2 hour and 1 hour before their habitual sleep onset for the participant with PD and healthy control, respectively. The arrow heads indicate habitual sleep onset which was not different between both participants.
The mechanisms through which circadian and sleep disruption occurs in PD is unclear. It was suggested that altered clock gene expression in the SCN or peripheral clocks (BMAL1, PER1) may be causal, with BMAL1 showing lower expression in PD [29, 30]. Direct damage to the SCN by Lewy body formation or decreased expression of melatonin receptors in the substantia nigra and amygdala are also thought to induce circadian and sleep disruption. However, no study has previously considered the ipRGC contribution to circadian and sleep disruption in PD. Studies in Opn4 knock out mice indicate that normally functioning ipRGCs are required to promote the full effect of sleep [9]. IpRGC M1 subtypes transmit light signals to various brain areas with direct inputs to the SCN (circadian photoentrainment), VLPO (sleep) as well as to the OPN for regulating the pupil light reflex [8]. Our findings of a reduced ipRGC-mediated pupil response (Fig. 2 h) suggests that photic inputs transmitted via dysfunctional ipRGCs to the SCN and sleep promoting centers (e.g., VLPO) are attenuated, thereby conveying irregular light-dark signals which leads to poorer sleep.
Dysfunctional ipRGC signaling might be linked to reduced dopaminergic neurotransmission in PD [31]. Dopamine attenuates the melanopsin light response by acting on D1-type receptors expressed in the dendrites of ipRGCs. In mice, M1 ipRGCs receive modulatory input (inhibitory) from dopamine amacrine (DA) cells and also have reciprocal signaling (excitatory) to DA cells [32]. This specific retrograde light-signaling pathway suggests a circuitry that links DA and ipRGC function and loss in DA neurons might potentially desynchronize the circadian system. The human M1 ipRGC subtype mainly receives synaptic inputs from dopaminergic axons and this M1 subtype undergoes morphological changes as demonstrated in human PD retinae [11].
Our results of a longer entrainment phase angle in people with PD is consistent with past research by Bolitho et al. [5]. In addition, we show an earlier melatonin onset that provides further evidence of desynchronized circadian entrainment in people with PD. A number of participants returned salivary DLMO data below assay detection levels and were labelled as low secretors. While this may be a limitation of salivatory DLMO determination, we note that our results of an earlier DLMO is in agreement with other studies in PD [5]. Salivary DLMO has many advantages over plasma melatonin determination that returns higher levels such as for example being non-invasive and requiring minimal sample handling [33].
A strength of our study is the measurement of environmental light over an extended 14-day period as opposed to previous studies that determined melatonin but did not account for ambient illumination [5]. There have been conflicting results regarding melatonin concentration and melatonin onset in PD [1, 5], and an increase in melatonin concentration and an entrainment phase advance have been mainly attributed to dopaminergic drugs [2, 5]. The latter can lead to increased melatonin secretion due to the β-adrenergic innervation of melatonin-producing pinealocytes that may impact on DLMO. We did not investigate this relationship as all our participants with PD were optimally medicated. Importantly, we did not observe a relationship between DLMO, phase angle and daily dopamine dosage and dopamine agonists, indicating that medication was not associated with the phase advance and earlier melatonin onset.
Light is effective for improving sleep, mood and motor symptoms in PD [34] but evidence-based data concerning the underlying mechanisms were lacking. Our findings provide a physiological rationale for developing new treatment strategies using supplemental artificial light exposure that targets melanopsin activation in people with PD to limit the impact of sleep disruption and improve quality of life.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
Footnotes
ACKNOWLEDGMENTS
This work was supported by the Australian Research Council (ARC-DP170100274, ARC-FT 180100458).