
Abstract
Background:
Increased firing across glutamatergic synapses may contribute to both the motor dysfunction and L-DOPA-induced dyskinesia seen in Parkinson’s disease. Given their ability to reduce glutamate release, activation of group III metabotropic glutamate receptors such as metabotropic glutamate receptor 4 may prove effective against both motor dysfunction and dyskinesia in Parkinson’s disease.
Objective:
We hypothesised that activation of metabotropic glutamate receptor 4 by an orthosteric agonist ((2S)-2-amino-4-(hydroxy(hydroxy(4-hydroxy-3-methoxy-5-nitrophenyl)methyl)phosphoryl)butanoic acid, LSP1-2111) would produce antiparkinsonian activity and reduce expression of dyskinesia in a 1-methyl-4-phenyl,1,2,3,6-tetrahydropyridine (MPTP)-treated marmoset model of Parkinson’s disease.
Methods:
Common marmosets were previously treated with MPTP and pre-primed with L-DOPA for up to 28 days to express dyskinesia. LSP1-2111 (1, 3, or 6 mg/kg s.c.) or vehicle (0.9% saline s.c.) were administered immediately prior to L-DOPA (8 mg/kg + benserazide (10 mg/kg) p.o.) or vehicle (10% sucrose p.o.). Locomotor activity was measured in automated test cages and animals were scored for dyskinesia and disability.
Results:
As expected, L-DOPA reversed motor disability and induced moderate dyskinesia. By contrast, LSP1-2111 alone significantly reduced the motor disability without any accompanying expression of dyskinesia. When administered in combination with L-DOPA, LSP1-2111 did not significantly reduce the severity of L-DOPA-induced dyskinesia.
Conclusion:
Systemic administration of LSP1-2111 reduces motor disability without causing dyskinesia in MPTP-treated marmosets, supporting a role for metabotropic glutamate receptor 4 orthosteric agonists as promising monotherapy for PD. Conversely, this study found no evidence to support their use as antidyskinetic agents within the dose range tested.
INTRODUCTION
Parkinson’s disease (PD) is a progressive neurodegenerative disorder that presents with motor (e.g., bradykinesia, tremor, and postural instability) and non-motor (e.g., pain, anxiety, and REM-sleep behaviour disorder) symptoms. The current gold standard treatment for PD is L-3,4-dihydroxyphenylalanine (L-DOPA), which provides relief from motor symptoms. However, within 4–6 years after the initiation of L-DOPA treatment, 40% of PD patients experience unwanted involuntary movements in the form of L-DOPA-induced dyskinesia (LID) of a choreic or dystonic nature [1].
Increased glutamatergic transmission has been implicated in the pathophysiology of both parkinsonian motor symptoms and L-DOPA-induced dyskinesia [2, 3]. Increased transmission across the glutamatergic subthalamonigral pathway is believed to contribute towards manifestation of the motor symptoms [4, 5], while plasticity of the glutamatergic corticostriatal pathway is implicated in the development of LID [6–9]. In support of the glutamatergic involvement in LID, the weak N-methyl-D-aspartate (NMDA) receptor antagonist amantadine is one of very few drugs shown to have any efficacy against LID [10–13]. However, amantadine has a poor side-effect profile involving psychiatric problems such as hallucination, confusion and depression [14] which reduces its therapeutic utility. An alternative route to the glutamatergic modulation of signalling for potential therapeutic benefit against both the parkinsonian motor symptoms and LID is to target metabotropic glutamate receptors, specifically, the group III metabotropic glutamate receptors (mGluRs) which have shown promise in a range of PD and LID indications [2, 15].
Group III mGluRs are Gi/o-coupled, presynaptic receptors which reduce exocytosis of neurotransmitter in response to activation by endogenous glutamate [16–18]. One member of this family, mGluR4, has received attention as a potential therapeutic target in PD due to its expression at relevant synapses throughout the basal ganglia [15, 19–21]. Indeed, both agonists and positive allosteric modulators (PAMs) of mGluR4 have been shown to provide antiparkinsonian effects in acute models of PD in rodents [22–25], while PAMs offer antiparkinsonian relief in a 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-treated macaque model of PD [26].
Studies have also shown the antidyskinetic potential of targeting mGluR4. Thus, the mGluR4 PAM, (1S, 2R)-N1-(3,4-dichlorophenyl)-cyclohexane-1,2-dicarboxamide (Lu AF219234), reduced the development of L-DOPA-induced abnormal involuntary movements (AIMs) in rodent models of LID [24]. Similarly, the mGluR4 PAM, Foliglurax (PXT002331), reduced the expression of well-established LID in the MPTP-treated macaques [26]. A systemically-active agonist of mGluR4, (2S)-2-amino-4-(hydroxy(hydroxy(4-hydroxy-3-methoxy-5-nitrophenyl)methyl)phosphoryl)butanoic acid (LSP1-2111) has also shown efficacy against the development of L-DOPA-induced AIMs in rodents [27]. However, whether an mGluR4 agonist will offer beneficial effects in a primate model of PD remains to be examined. This study therefore set out to establish whether the mGluR4 agonist LSP1-2111 provides antiparkinsonian relief in an MPTP-treated marmoset model of PD and whether it also reduces the expression of established LID in this model.
MATERIALS AND METHODS
Animals
Common marmosets (Callithrix jacchus, Harlan, Loughborough, LE12 9TE, UK and Manchester University, UK) aged 7–14 years were housed in female/male (vasectomised) or female/female pairs at a temperature of 23±2°C with 50% relative humidity and a 12 h light/dark cycle [28, 29]. They had unlimited access to water and marmoset pellets and received one meal of mashed cereal and one meal of fresh fruit daily. All experiments were performed according to the Animals (Scientific Procedures Act) 1986 under Project Licence No 70/8541, with local approval of the Animal Welfare and Ethical Review Board of King’s College London and were compliant with the minimum standards as defined by the European Communities Council Directive (10/63/EU). All animals involved in this study had previously been included in studies assessing the therapeutic value of compounds in PD and LID. Following previous studies, all animals underwent a drug-free ‘washout’ period of at least 4 weeks before the start of this study.
MPTP treatment
Five to seven years prior to this study, marmosets underwent administration of MPTP (Sigma, UK) at 2.0 mg/kg daily for 5 days to induce stable motor deficits [30, 31]. This resulted in the animals exhibiting reduced basal locomotor activity, bradykinesia, rigidity, poor coordination of movement and reduced alertness/awareness. All animals were primed to express dyskinesia on exposure to L-DOPA through repeated (up to 28 days) oral administration of L-DOPA (8-12.5 mg/kg, Sigma, UK) plus benserazide (10 mg/kg, Sigma, UK) in a 10% sucrose solution.
Drug treatment
Animals (n = 8) were selected for the study from a pool (n = 10) of MPTP-treated marmosets based on their response to L-DOPA treatment. For this selection process L-DOPA (4, 6, and 8 mg/kg) plus benserazide (10 mg/kg) was administered p.o. and locomotor activity recorded (as detailed below). L-DOPA (8 mg/kg) was selected as the dose (providing approximately 70% of a maximal response) for use in the main part of the study. Two animals were removed from the study prior to completion for welfare reasons unrelated to the study, leaving a final group size of n = 6 (3 male and 3 female).
A modified Latin square design was used to randomise treatments whilst ensuring dosing with LSP1-2111 (Lundbeck, Denmark) occurred in a dose-escalating manner, to identify any side-effects before higher doses were given. In this fashion, each animal received all drug combinations once (with an interval of ≥48 h between doses). LSP1-2111 was administered subcutaneously in 0.9% sterile saline (Baxter healthcare) at 0 (vehicle) 1, 3, or 6 mg/kg in a volume of 1 ml/kg. The lowest dose for LSP1-2111 (1 mg/kg) was selected based on previous data showing emerging significant effects in a range of behavioural tests in rodents with this dose [25, 32].
Following a 60 min acclimatisation period, baseline motor function (locomotor activity, motor disability and dyskinesia) was assessed for 60 min as described below. Following the 60 min baseline assessment, LSP1-2111 and L-DOPA (or respective vehicles) were administered according to the randomisation protocol. LSP1-2111 (1, 3, or 6 mg/kg s.c.) or vehicle (1 ml/kg s.c.) was administered immediately followed by L-DOPA (8 mg/kg plus benserazide (10 mg/kg)) or vehicle (10% sucrose plus benserazide (10 mg/kg)) in a combined p.o. administration of 2 ml/kg.
Behavioural measurements
On test days, animals were acclimatised for 60 min to individual automated test units (50 cm by 60 cm by 90 cm). The automated test units were fitted with 2 horizontal wooden perches and a water supply and a clear Perspex door to allow visual observation. Food was not provided during the test period and animals received their normal meal at the end of the test period on return to home caging. Locomotor activity, motor disability, and dyskinesia were assessed for up to 6 h as described below.
Locomotor activity
Each behavioural test unit was fitted with 8 photoelectric emitters/detectors (light beams) arranged horizontally to permit optimal assessment of locomotor activity. Interruption of a light beam was automatically recorded as a single locomotor count which were accumulated in 30 min time segments for 1 h before and 5 h following drug treatment.
Motor disability
Motor disability was assessed simultaneously with locomotor activity, by observation via a one-way mirror, by experienced observers blinded to the treatment. Basal disability was assessed once every 30 min, for 30 min before and 5 h after drug treatment using an established motor disability rating scale; alertness (normal = 0, reduced = 1, sleepy = 2); checking (present = 0, reduced = 1, absent = 2); posture (normal = 0, abnormal trunk +1, abnormal tail +1, abnormal limbs +1, flexed = 4); balance (normal = 0, impaired = 1, unstable = 2, spontaneous falls = 3); reaction to stimuli (normal = 0, reduced = 1, slow = 2, absent = 3); vocalisation (normal = 0, reduced = 1, absent = 2); motility (normal = 0, bradykinesia = 1, akinesia = 2). These values were summed, a maximum score of 18 indicating severe motor disability, a minimum score of 0 indicating maximum reversal of motor disability.
Dyskinesia
Dyskinesia was assessed simultaneously with motor disability by experienced observers blinded to treatment. The following established dyskinesia rating scale was used; 0 = absent; 1 = mild, fleeting and rare dyskinetic postures and movements; 2 = moderate: more prominent abnormal movements, but not significantly affecting normal behaviour; 3 = marked, frequent and at times continuous dyskinesia affecting the normal pattern of activity; 4 = severe, virtually continuous dyskinetic activity, disabling to the animal and replacing normal behaviour.
Data handling and statistical analysis
The area under the curve (AUC) for locomotor activity, motor disability and dyskinesia was determined from the time course data over 5 h following drug administration (GraphPad Prism version 8.0.0 for Windows, GraphPad Software, San Diego California USA, http://www.graphpad.com). The AUC for locomotor activity and dyskinesia was calculated from values greater than baseline and for reversal of motor disability values lower than baseline. For AUC figures therefore, increased locomotor activity, reversal of motor disability and increased severity of dyskinesia are all represented by rising values.
Prior to analysis, locomotor activity, motor disability and dyskinesia data were transformed by y =√y in order to normalise distribution [33]. This transformation allowed the application of parametric tests to scored data. Time course data was analyses by 2-way ANOVA. If the effect of treatment was significant, individual differences at each time point were analysed by Holm Sidak’s test. Repeated measures 1-way ANOVA with Sidak’s multiple comparisons test was applied to area under the curve (AUC) data, comparing each group to its respective control.
RESULTS
Vehicle treatment had no effect on either locomotor activity or motor disability and did not induce dyskinesia expression (Figs. 1–3).
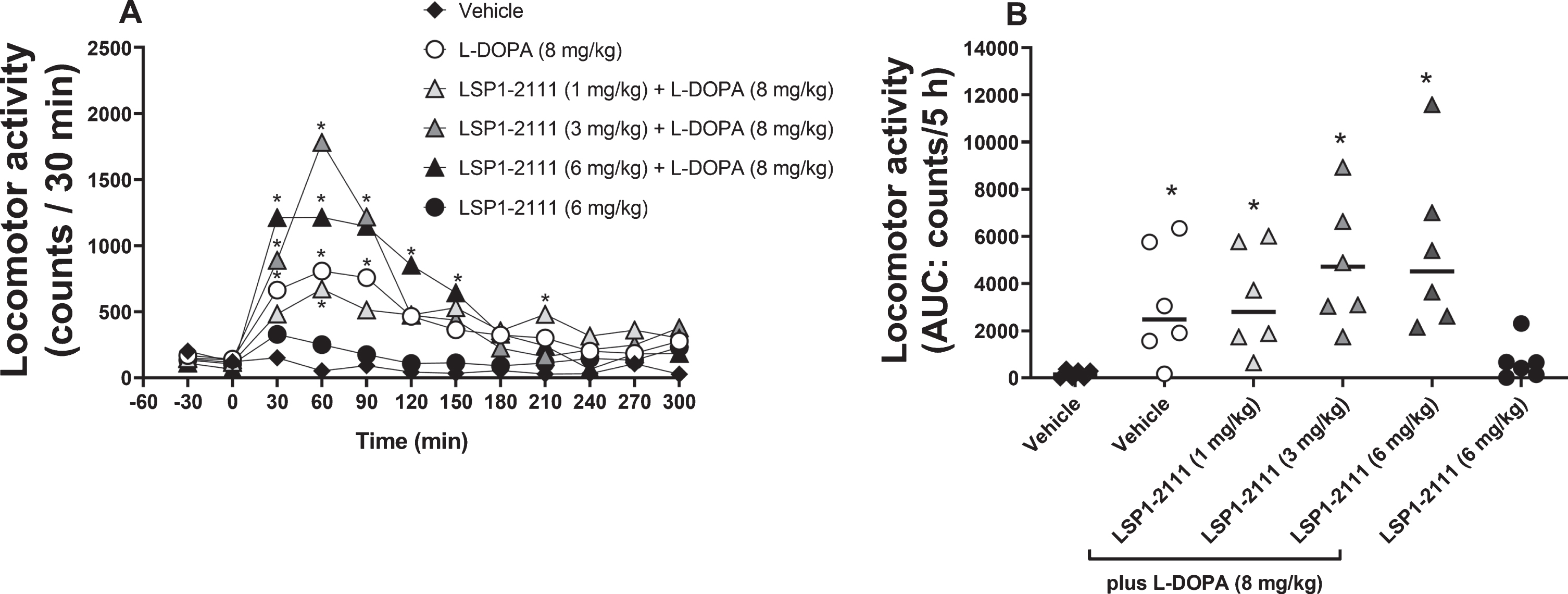
The effect of treatment with ((2S)-2-amino-4-(hydroxy(hydroxy(4-hydroxy-3-methoxy-5-nitrophenyl)methyl)phosphoryl)butanoic acid (LSP1-2111; 6 mg/kg p.o.) alone or with L-DOPA in the presence of increasing doses of LSP1-2111 (vehicle, 1, 3, and 6 mg/kg p.o.) on locomotor activity. A) The time course of effect with treatment administered at time T = 0. Data are presented as mean locomotor counts per 30 min (n = 6). *p < 0.05 versus vehicle alone (two-way ANOVA plus Holm-Sidak’s multiple comparison test on transformed data). B) Total locomotor activity counts (AUC). Data are presented as mean (line) and individual counts. *p < 0.05 versus vehicle alone (♦) (one-way ANOVA plus Holm-Sidak’s multiple comparison test on transformed data).
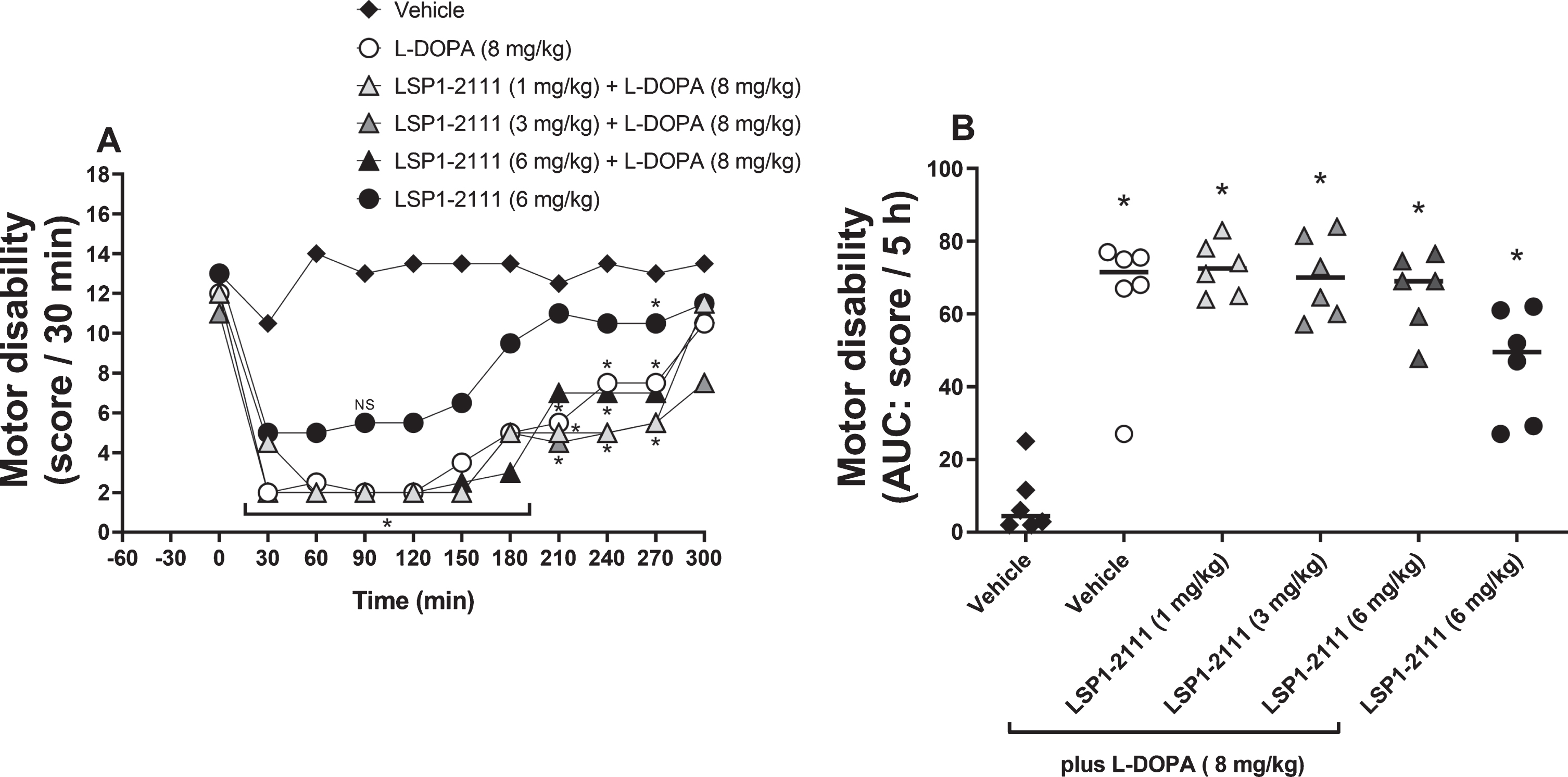
The effect of treatment with ((2S)-2-amino-4-(hydroxy(hydroxy(4-hydroxy-3-methoxy-5-nitrophenyl)methyl)phosphoryl)butanoic acid (LSP1-2111; 6 mg/kg p.o.) alone or with L-DOPA in the presence of increasing doses of LSP1-2111 (vehicle, 1, 3, and 6 mg/kg p.o.) on motor disability. A) The time course of effect with treatment administered at time T = 0. Data are presented as median scores per 30 min (n = 6). *p < 0.05 all groups versus vehicle alone, NS indicates single point of non-significance (two-way ANOVA plus Holm-Sidak’s multiple comparison test on transformed data). B) Total reversal of motor disability (AUC). Data are presented as median (line) and individual counts. *p < 0.05 versus vehicle alone (♦) (one-way ANOVA plus Holm-Sidak’s multiple comparison test on transformed data).
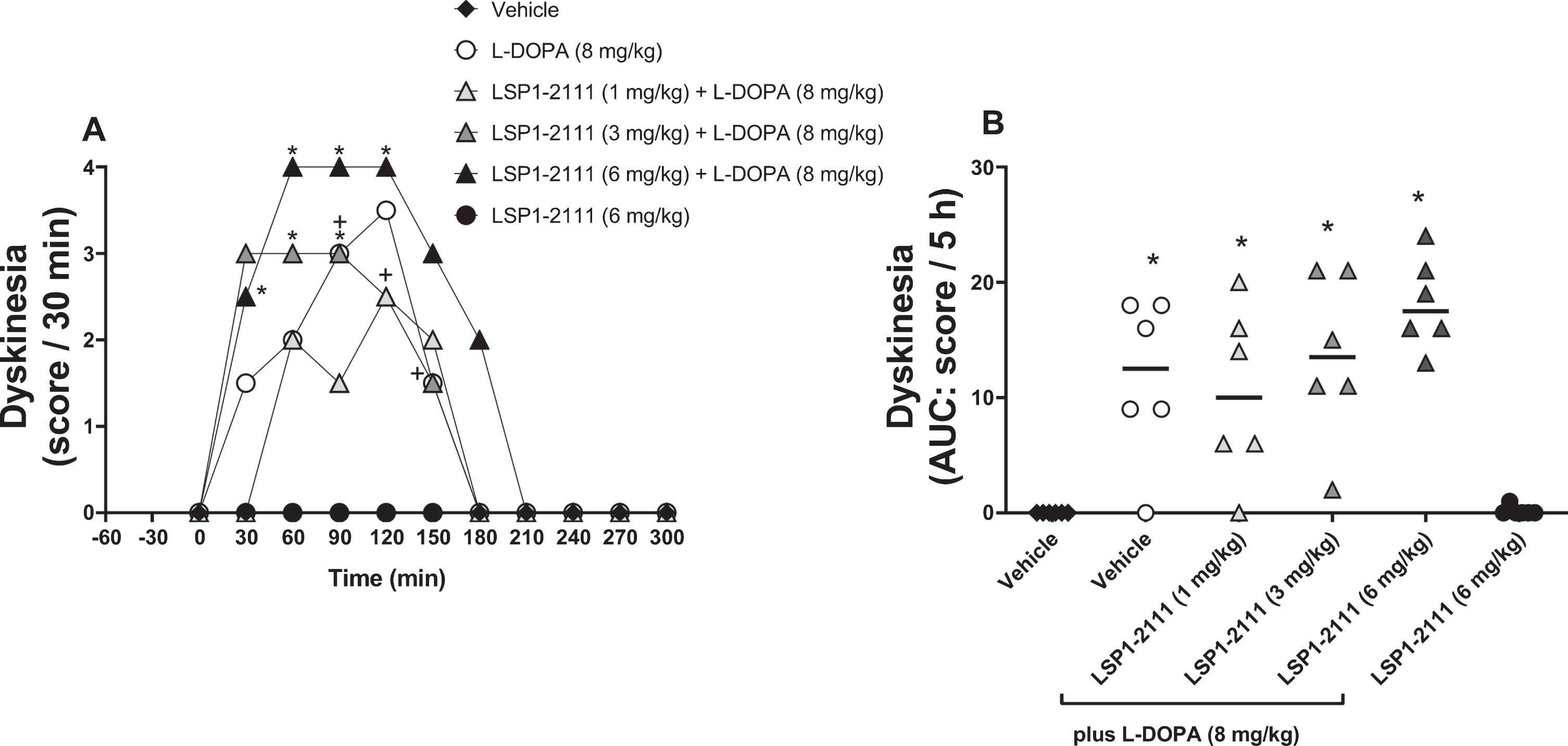
The effect of treatment with ((2S)-2-amino-4-(hydroxy(hydroxy(4-hydroxy-3-methoxy-5-nitrophenyl)methyl)phosphoryl)butanoic acid (LSP1-2111; 6 mg/kg p.o.) alone or with L-DOPA in the presence of increasing doses of LSP1-2111 (vehicle, 1, 3, and 6 mg/kg p.o.) on dyskinesia expression. A) The time course of effect with treatment administered at time T = 0. Data are presented as median scores per 30 min (n = 6).+p < 0.05 vehicle versus L-DOPA alone, *p < 0.05 vehicle versus L-DOPA plus LSP1-2111 combinations (two-way ANOVA plus Holm-Sidak’s multiple comparison test on transformed data). B) Total dyskinesia score (AUC). Data are presented as median (line) and individual counts. *p < 0.05 versus vehicle alone (♦) (one-way ANOVA plus Holm-Sidak’s multiple comparison test on transformed data).
As expected, the submaximal dose of L-DOPA (8 mg/kg p.o.) produced a small but significant rise in locomotor activity (Fig. 1a,b), a significant reversal of motor disability (Fig. 2a,b) and significant expression of dyskinesia (Fig. 3a,b). Locomotor activity peaked at 60 min (Fig. 1a), whilst the improvement in motor disability showed maximum effect between 30 and 90 min after administration (Fig. 2a) with scores of 2. Dyskinesia peaked between 90 and 120 min with moderate to marked dyskinetic movements (median scores of 2–3). This effect of L-DOPA lasted approximately 3 h.
LSP1-2111 alone (6 mg/kg s.c.) had no effect on locomotor activity (Fig. 1a,b) but significantly improved motor disability with a sub-maximal reduction in score between 30 and 60 min (Fig. 2a,b). Interestingly, LSP1-2111 (6 mg/kg s.c.) did not induce any dyskinesia (Fig. 3a,b).
When given in combination with L-DOPA (8 mg/kg p.o.), LSP1-2111 (1–6 mg/kg) appeared to increase locomotor activity in a dose-related manner, although this effect was not significant (Fig. 1a,b). In spite of the reversal of motor disability by LSP1-2111 (6 mg/kg s.c.) alone, when given in combination, LSP1-2111 (1–6 mg/kg s.c.) did not alter the L-DOPA-induced reversal of motor disability (Fig. 2a,b). However, in parallel with the non-significant rise in locomotor activity, LSP1-2111 produced a non-significant increase in the expression of L-DOPA-induced dyskinesia at the highest dose tested (Fig. 3a,b). This included a dose-related increase in chorea, but not dystonia (Supplementary Figure 1) with fleeting bouts of severe choreic activity at peak effect after the combination of LSP1-2111 (6 mg/kg) and L-DOPA. For this reason, the effects of further increments in dose of LSP1-2111 were not explored.
DISCUSSION
This study set out to examine whether the mGluR4 agonist, LSP1-2111, provided antiparkinsonian relief or reduced the expression of established LID in the MPTP-treated marmoset. LSP1-2111 alone was shown to significantly reduce motor disability in parkinsonian animals without causing dyskinesia. However, LSP1-2111 did not reduce established LID when co-administered with L-DOPA.
Regarding the potential antiparkinsonian efficacy of LSP1-2111, the significant reduction in motor disability seen with LSP1-2111 alone compared to vehicle treatment supports an antiparkinsonian effect of this mGluR4 agonist. Although the reduction in motor disability was non-significantly lower than that achieved with L-DOPA treatment, these animals were clearly ‘switched on’ as defined by a score of 8 [34]. Importantly, in contrast to the response with L-DOPA, this beneficial effect of LSP-2111 was not accompanied by a significant increase in locomotor activity, indicating less hyperactivity, and more naturalistic antiparkinsonian effect. Furthermore, administration of LSP1-2111 alone did not evoke the expression of dyskinesia in L-DOPA-primed animals.
LSP1-2111 did not have any significant additive effects in reversing motor disability when given alongside the submaximal dose of L-DOPA (8 mg/kg) used here. This suggests that the LSP1-2111 operates via the same downstream mechanism as L-DOPA to achieve this antiparkinsonian response. Existing evidence points towards a mechanism involving modulation of the indirect pathway of the basal ganglia to counteract pathological alterations in firing. For example, in vitro slice work has shown that activation of mGluR4 receptors, using either agonists or PAMs, reduces GABAergic transmission across the striatopallidal pathway, reflecting the heteroreceptor role of these receptors [35–37] and glutamatergic transmission across the subthalamonigral [22, 38] and corticostriatal [24, 37] pathways, reflecting the autoreceptor roles. The outcome of each of these actions is to reduce the overall activity in the indirect pathway, restoring the balance of firing between the direct and indirect pathways which is thought to be disrupted in PD [39, 40], thereby restoring motor function.
In contrast to the antiparkinsonian effect of LSP1-2111 noted here, treatment with the mGluR4 PAM, PXT002331, did not elicit a robust antiparkinsonian effect when given alone to MPTP-treated macaques modelling either early or late stage PD [26]. While this may reflect differences between the macaque and marmoset models of PD, a more likely explanation is that mGluR4 agonists provide greater activation of the relevant receptors. To activate mGluR4, an orthosteric agonist like LSP1-2111 does not require additional endogenous glutamate. However, a PAM such as PXT002331 requires the presence of endogenous glutamate to stimulate the orthosteric site, before the action of the PAM is manifest. Although sufficient glutamate might be anticipated at the corticostriatal and subthalamonigral synapses to support actions of a PAM, this is unlikely to be so at the GABAergic striatopallidal synapse. Therefore, one possible explanation why the mGluR4 agonist but not PAM is antiparkinsonian when administered alone, is that the additional activity of the agonist at the striatopallidal synapse is key to underpinning the antiparkinsonian efficacy.
Although not effective when administered alone, the mGluR4 PAM, PTX002331, did enhance the locomotor response to L-DOPA [26] and this L-DOPA sparing action was also not accompanied by the emergence of dyskinesia. In partial agreement with this, in the present study LSP1-2111 tended to enhance the locomotor activity AUC with L-DOPA from 3134±999 counts/5 h (L-DOPA alone) to 5395±1440 counts/5 h (L-DOPA plus 6 mg/kg LSP1-2111) although this failed to reach significance. However, a L-DOPA sparing action per se was not examined in this study. This would have required administering LSP1-2111 with a subthreshold dose of L-DOPA. Given our primary aim was to explore the anti-dyskinetic effect of LSP1-2111, it was only given here alongside suprathreshold doses of L-DOPA that elicited significant dyskinesia. Nevertheless, our data provide support for mGluR4 agonists being more effective than PAMs as a monotherapy, while PAMs may prove more effective as an adjunct to L-DOPA.
A second aim of this study was to examine the potential of LSP1-2111 to reduce LID in MPTP-treated marmosets. The present data clearly show that LSP1-2111 has no antidyskinetic effect. At all doses tested, co-administration of LSP1-2111 failed to reduce the extent of LID compared to that evoked by administration of L-DOPA alone. Rather, LSP1-2111 tended to increase the expression of LID from a median AUC score/5 h of 12.5 (range 18; L-DOPA alone) to 17.5 (range 11; L-DOPA plus 6 mg/kg LSP1-2111). Although this failed to reach significance, a dose-related increase in choreic movements was observed, and prevented higher doses being tested. This lack of antidyskinetic effect agrees with previous studies in rodents which also found no beneficial effect of a single administration of LSP1-2111 to animals with pre-established dyskinesia [25, 27]. Given that plasticity across the corticostriatal synapse is central to the pathophysiology of LID [6–9], the lack of antidyskinetic efficacy with LSP1-2111 suggests that modulation across this synapse using an mGluR4 agonist is not likely to have a functional outcome in vivo. Accordingly, this also points to effects at either the striatopallidal synapse (as previously discussed) or subthalamonigral synapse underlying the above antiparkinsonian actions of LSP1-2111, rather than an action on the corticostriatal synapse.
In contrast to the lack of antidyskinetic efficacy with LSP1-2111, the single published primate study with an mGluR4 PAM (PTX002331) did reveal an antidyskinetic effect in a macaque model of late stage PD expressing established LID [26]. The reason behind these different outcomes with agonist versus PAM remains to be established. One possibility is that a modulatory action on the relevant mGluR4 receptors—most likely those at the corticostriatal synapse for dyskinesia—is more likely to normalise firing levels compared to outright activation with an agonist which might instead lead to too much inhibition of glutamate release and excessively reduced firing in downstream pathways. An alternative explanation is that the mGluR4 agonist may act at multiple sites in the striatum that counteract each other. For example, a related mGluR4 agonist, LSP1-3081 has been shown to inhibit GABA release in the striatum, as well as glutamate release [41]. If LSP1-2111 acts similarly on heteroreceptors to reduce GABA release in the striatum, this could counter any potential antidyskinetic efficacy of LSP1-2111’s action on the corticostriatal pathway. Depending on the location of these heteroreceptors, it is plausible that they are not modulated by an mGluR4 PAM, due to a lack of sufficient glutamate at the orthosteric site, permitting the antidyskinetic effects of the PAM to prevail.
When administered to rodents in combination with L-DOPA, LSP1-2111 did reduce LID induction in one [27] but not another [25] study. It will therefore be important in the future to determine whether this mGluR4 agonist can reduce the incidence or severity of LID when given in combination with L-DOPA in de novo treated marmosets. Such an outcome is also not yet known for mGluR4 PAMs.
One potential disadvantage of mGluR4 agonists over PAMs that requires consideration is the risk of triggering receptor desensitisation with chronic use. However, given that chronic administration of LSP1-2111 was efficacious in a rodent model of LID [27], it seems that desensitisation may not be of concern with this agonist. Indeed, studies have shown that desensitisation of mGluR4 is independent of agonist activation [42], thus mGluR4 agonists remain serious contenders for use in PD.
Summary
In summary, this study is the first to examine the antiparkinsonian and antidyskinetic efficacy of an mGluR4 agonist in a primate model of PD. Although unable to reduce the severity of established LID, our data reveal that LSP1-2111 produces an anti-parkinsonian effect, without provoking dyskinesia in L-DOPA primed MPTP-treated marmosets, supporting further examination of the potential of mGluR4 agonists in the treatment of PD.
CONFLICT OF INTEREST
The authors report no conflicts of interest in relation to the content of this manuscript.