
Abstract
Background:
Accumulation of α-synuclein (αSyn) in the dopaminergic neurons is a common pathology seen in patients with Parkinson’s disease (PD). Overproduction of αSyn potentiates the formation of oligomeric αSyn aggregates and enhances dopaminergic neuron degeneration. Downregulating intracellular monomeric αSyn prevents the formation of αSyn oligomers and is a potential therapeutic strategy to attenuate the progression of PD.
Objective:
The purpose of this study is to investigate the efficacy of gene delivery of αSyn-specific single-chain antibodies in vitro and in vivo.
Methods and Results:
The plasmids for αSyn and selective antibodies (NAC32, D10, and VH14) were constructed and were transfected to HEK293 and SH-SY5Y cells. Co-expression of αSyn with NAC32, but not D10 or VH14, profoundly downregulated αSyn protein, but not αSyn mRNA levels in these cells. The interaction of αSyn and NAC32 antibody was next examined in vivo. Adeno-associated virus (AAV)-αSyn combined with AAV-NAC32 or AAV-sc6H4 (a negative control virus) were stereotactically injected into the substantia nigra of adult rats. AAV-NAC32 significantly reduced AAV-encoded αSyn levels in the substantia nigra and striatum and increased tyrosine hydroxylase immunoreactivity in the striatum. Also, in the animals injected with AAV-NAC32 alone, endogenous αSyn protein levels were significantly downregulated in the substantia nigra.
Conclusion:
Our data suggest that AAV-mediated gene transfer of NAC32 is a feasible approach for reducing the expression of target αSyn protein in brain.
INTRODUCTION
Parkinson’s disease (PD) is the second most common neurodegenerative disease after Alzheimer’s disease and affects 1–2% of individuals over the age of 65 [1]. PD is characterized by a series of motor symptoms including slowness of movement (bradykinesia), resting tremor, muscular rigidity, and postural instability, due to the progressive loss of dopaminergic neurons in the midbrain substantia nigra pars compacta (SNpc) [2, 3]. Current treatments for PD mainly rely on pharmacological strategies that provide symptomatic relief of motor dysfunctions. For example, L-DOPA increases dopamine production in the remaining dopaminergic neurons [4]. However, symptom improvement during L-DOPA therapy does not alter the progression of neurodegeneration in dopaminergic neurons. Its effectiveness is also limited by several adverse effects, such as the development of dyskinesia, which negatively affect patients’ quality of life. Therefore, development of novel therapeutic strategies for PD is needed and warrants further investigation.
The cardinal neuropathological features of PD include the progressive loss of dopaminergic neurons and the accumulation of misfolded α-synuclein (αSyn) in the cytoplasm of remaining dopaminergic neurons [5]. Several separate point mutations in the αSyn gene (i.e., A30P, E46K, H50Q, G51D, and A53T), as well as duplication and triplication of wild type αSyn, have been identified in patients with familial forms of early-onset PD. In addition, genome-wide association studies have revealed the αSyn gene as a risk locus for the sporadic PD, the most common variant [6–8]. Although the mechanism of αSyn-mediated dopaminergic cell death is not fully understood, over-expression of αSyn in rodents and drosophila induces progressive neurodegeneration and motor impairments similar to PD in patients [9–11]. However, there are many more studies published with AAV-αSyn models, and not all of the conclusions are that straightforward [12–15]. αSyn is a neuronal protein of 140 amino acids that is abundantly expressed in presynaptic terminals, with a possible function in the regulation of neurotransmitter release [16, 17]. The native protein is unfolded and has three distinct regions: (1) an amphipathic N-terminus, which can form α-helical structures on binding to cellular membranes, (2) a central hydrophobic region (61–95 residues) termed the non-amyloid-beta component (NAC), which is aggregation-prone, (3) and a highly negatively charged C-terminus, which has no defined known structure [18].
Excessive αSyn potentiates misfolding, aggregation, and formation of oligomeric intermediate species as well as insoluble fibrillar inclusions under stress conditions, and the oligomeric form of αSyn has been suggested to be neurotoxic [19, 20]. Abnormal accumulation of oligomeric αSyn in axons and presynaptic terminals is critical in the process of αSyn-mediated neurodegeneration. Therefore, reducing intracellular monomeric αSyn levels to prevent the formation of αSyn oligomers or to clear existing oligomers in affected neurons can be a valuable strategy to attenuate toxic insults and slow down the progression of PD. Lewy bodies (LBs) are intracellular deposits of eosinophilic protein aggregates that are primarily composed of αSyn proteins. In the clinic, neither the distribution nor density of LBs was associated with the severity of dopaminergic cell loss in the substantia nigra [21, 22]. Despite this discrepancy, αSyn aggregation is clearly playing a role in disease progression in PD patients. Thus, downregulation of intracellular αSyn protein levels remains a valid strategy for PD treatment.
The immunoglobulin G (IgG) antibodies are secreted glycoproteins produced by plasma cells that terminally differentiate from antigen-stimulated B lymphocytes in the immune system. The IgG antibodies of mammalian species are built from two identical heavy-chain (consisting of a variable and three constant domains; VH, CH1, CH2, CH3) and two identical light-chain (composed of a variable and a constant domain; VL, CL) proteins, which assemble into a Y-shape structure. The antibody contains two identical antigen-binding sites, each formed by the paired variable domains of heavy and light chains [23]. The Camelidae species produces not only conventional IgG antibodies but also a considerable amount of heavy-chain antibodies (HCAbs), which consist of two identical heavy-chain proteins but lack light-chain proteins [24]. The heavy chain within HCAbs also lacks the first constant domain (CH1), and the amino acid sequence of the remaining constant domain (CH2-CH3) is highly similar to that of conventional antibodies [25]. The heavy-chain variable domain of HCAbs, referred to as VHH or nanobody, is functional in antigen binding without the requirement of pairing with a light-chain variable domain [26, 27]. Intrabodies are single-chain variable fragments (scFv; a VH connected to a VL by a peptide), or single-domain fragments (VH or VL) of conventional antibodies or VHHs of HCAbs expressed intracellularly. In contrast to secreted full-length antibodies, which target extracellular antigens, intrabodies target antigens inside the cell [28, 29]. Due to the reducing redox potential disrupting disulfide bonds in the cytoplasm and absence of endoplasmic reticulum chaperones, intrabodies expressed in the cytosol tend to fold incorrectly [30, 31]. However, many intracellularly expressed intrabodies have been demonstrated to retain affinity and specificity for the corresponding target proteins. Upon binding to their target proteins, intrabodies may induce several effects on the bound target through disrupting interactions with other partner components, altering the stability, or promoting degradation.
Numerous intrabodies have been successfully used to promote tumor regression via blocking intracellular signaling networks of oncogenic proteins such as Ras and erbB-2 [32–34]. Intracellular expression of intrabodies against human immunodeficiency virus (HIV) proteins including gp120, reverse transcriptase, and Rev regulatory protein can lead to a profound reduction of HIV replication [35–37]. Intrabodies can also serve as therapeutics for neurodegenerative disorders that are associated with misfolded and accumulated intracellular proteins. An intrabody that selectively targets the polyglutamine residues of huntingtin can reduce huntingtin protein aggregation in cellular and organotypic slice culture models of Huntington’s disease (HD) [38, 39]. In addition, transgenic expression of this intrabody successfully delayed neurodegeneration and improved survival in an HD Drosophila model [40]. Intrabodies specifically binding to the β-secretase cleavage site in the β-amyloid precursor protein were shown to abolish the production of amyloid b-peptide in a cellular model of Alzheimer’s disease [41]. An intrabody against monomeric αSyn has been demonstrated to inhibit the formation of oligomeric αSyn and ameliorate cell-adhesion defects induced by αSyn over-expression in a cellular model of Parkinson’s disease [42].
The present study was undertaken to examine the effect of three αSyn-specific intrabodies on αSyn protein levels. We found that co-expression of αSyn and an intrabody targeting the NAC region of αSyn profoundly reduced αSyn protein levels in cultured cells and rat brain. Our data encourage further studies to investigate the therapeutic potential of intrabody-based gene therapy in an αSyn-overexpression rat model of PD.
MATERIALS AND METHODS
Cell lines
The human embryonic kidney 293 cell line (HEK293; #240073, Agilent Technologies) and the human neuroblastoma SH-SY5Y cell line (CRL-2266, ATCC) were cultured as monolayers in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% (v/v; 15% for SH-SY5Y cells) heat-inactivated fetal calf bovine serum (FBS), penicillin (100 IU/ml), and streptomycin (100 μg/ml) at 37°C in a humidified incubator with 5% CO2.
Plasmid construction
The gene for human αSyn, fused with a V5 tag at the 5’ end, was cloned into an adeno-associated virus shuttle plasmid at KpnI and EcoRV sites, resulting in the construct pAAV-αSyn. The cDNA sequences that encode a single-chain variable fragment antibody (scFv) against the methamphetamine molecule (sc6H4) [43], an intracellular scFv against the non-amyloid component (NAC) region of αSyn (NAC32) [44], an intracellular scFv against monomeric αSyn (D10) [42], and an intracellular single domain antibody against the NAC region of αSyn (VH14) [44] were synthesized by Genomics Ltd, Taiwan and then cloned into an adeno-associated virus shuttle plasmid pAAV-MCS (Agilent Technologies) at EcoRI and BamHI sites. The cDNA sequence encoding the FLAG tag was added at the 5’-end of these scFv genes mentioned above; the sequence of a post-transcriptional regulatory element derived from the woodchuck hepatitis B virus (WPRE; GeneBank accession no. J04514) was added at the 3’-end of these scFv genes to enhance mRNA stability and protein production. The fidelity of the cloned cDNA sequences on the final plasmids (pAAV-sc6H4, pAAV-NAC32, pAAV-D10, and pAAV-VH14) was confirmed by Sanger sequencing analysis by Genomics Ltd, Taiwan. The amino acid sequences, GenBank accession numbers, and reference information for the intrabodies used in this study are listed in Table 1.
Amino acid sequences and reference information of intrabodies
αSyn, alpha synuclein; Meth, methamphetamine; Ref, references.
Western blot analysis
HEK293 or SH-SY5Y cells were grown in 24-well plates (2×105 cells/well) overnight and then transfected with or without the plasmids (0.5 μg each) encoding the protein of interest, using jetPEI (#101-10N, Polyplus) transfection reagents. At 48 h post-transfection, samples were prepared for western blotting analysis. For the preparation of whole cell lysates, cells in each well were washed once with PBS and lysed in 100 μl of 2× Laemmli buffer (4% SDS, 125 mM Tris-HCl pH6.8, 10% β-mercaptoethanol, 20% glycerol, and 0.004% bromophenol blue in distilled water), and whole cell lysates were passed through a 25-gauge needle several times to reduce viscosity. For examining protein solubility, cells in each well were incubated with 100 μl of RIPA lysis buffer (#20-188, Millipore) on ice for 15 min; the lysate was homogenized by passing through a pipette several times and then centrifuged at 12,000×g at 4°C for 15 min. For examining rat brain samples, each collected brain tissue was homogenized in 100 μl of RIPA lysis buffer by vibration for 30 sec twice using a bead mill homogenizer at 4°C and then centrifuged at 12,000×g at 4°C for 15 min. The supernatant was mixed with 100 μl of 2× Laemmli buffer, and the pellet was washed with PBS three times and then resuspended with 50 μl of 2× Laemmli buffer. Each sample was heated at 90°C for 10 min and subjected to electrophoresis separation on 12% SDS-polyacryamide gels. The separated proteins were electrophoretically transferred from the gel to a PVDF membrane (#RPN303F, GE Healthcare). The membrane was then incubated with blocking buffer (5% skim milk and 0.1% Tween-20 in PBS) for 1 h and then incubated with a target-specific mouse monoclonal antibody against cellular beta-actin (1 : 20,000; #MAB1501, Millipore), V5 tag (1 : 2,000; #R960-25, Thermo), FLAG tag (1 : 500; #GTX629631, GeneTex), tyrosine hydroxylase (1 : 1,000; #AB152, Millipore), or endogenous αSyn (1 : 500; #610787, BD) in 10 ml blocking buffer for 2 h with gentle agitation. After washing with 0.1% Tween-20 (in PBS) three times for 10 min each, the membrane was incubated with a horseradish peroxidase (HRP)-conjugated goat polyclonal antibody (1 : 5,000; #GTX213111-01, GeneTex) against mouse IgG in 10 ml blocking buffer for 1 h, followed by the washing procedure described above. The light emission signals of the target proteins on the membrane were generated by using an enhanced chemiluminescence reagent (#RPN2106, GE Healthcare) and detected on X-ray films (#NEF596, Kodak). The intensity of the generated image on the film was quantified by ImageJ software (http://imagej.gov/ij).
RNA extraction
HEK293 cells were grown in 24-well plates (2×105 cells/well) overnight and then transfected with plasmids (0.5 μg each) by using jetPEI reagents. At 48 h post-transfection, cells were subjected to RNA extraction by Trizol Reagent (#15596-018, Ambion) according to the manufacturer’s instructions. Briefly, cells in each well were lysed with 0.4 ml of Trizol Reagent and the cell lysates were homogenized by pipetting up and down several times. After incubation on ice for 5 min, 80 μl of chloroform was added to the lysate, followed by incubation on ice for 3 min and then by centrifugation at 12,000×g at 4°C for 15 min. The upper aqueous phase containing the RNA was transferred to a 1.5 ml microcentrifuge tube and mixed with 0.2 ml of isopropanol followed by incubation on ice for 10 min and then by centrifugation at 12,000×g at 4°C for 10 min. The precipitated RNA pellet was washed with 0.4 ml of 75% ethanol, air-dried for 10 min, and resuspended in 30 μl of RNase-free water (#10977-015, Invitrogen). The resuspended RNA was treated with 2 unites of DNase I (#M0303 S, NEB) in a final volume of 100 μl 1×DNase I reaction buffer at 37°C for 10 min. RNA was then re-extracted by chloroform, re-precipitated by isopropanol, and washed by ethanol, as described above. Finally, the purified RNA was quantified by a NanoDrop spectrophotometer and stored at –80°C until use.
Quantitative real-time reverse transcription polymerase chain reaction analysis
The mRNA levels of αSyn in the HEK293 cells transfected with plasmids were determined by quantitative real-time reverse transcription polymerase chain reaction analysis (qRT-PCR) performed on an ABI Step One Plus system. First, the purified total RNA was subjected to reverse transcription for cDNA synthesis using Revert Aid First Stand cDNA Synthesis Kit (#K1622, Thermo Fisher Scientific). Briefly, 1 μg of RNA was mixed with 1 μl of random hexamer primers (100 μM) in distilled water to a volume of 12 μl and then incubated at 65°C for 5 min, followed by chilling on ice. Next, 4 μl of 5×reaction buffer, 1 μl of RNase inhibitor (20 U/μl), 2 μl of dNTP (10 mM), and 1 μl of M-MuLV reverse transcriptase (200 U/μl) were added to the chilled mixtures. The total mixture (20 μl) was incubated at 25°C for 5 min followed by 42°C for 60 min, and finally the reaction was terminated by heating at 70°C for 5 min. A 40-fold dilution of the resulting cDNA was made in distilled water. For the real-time PCR, 2 μl of diluted cDNA, 10 μl of 2× SYBR green PCR master mix (#K0371, Thermo Scientific), and 2 μl of each primer (5 μM) were mixed in distilled water to a final volume of 20 μl. The PCR cycling program was set as follows: 50°C for 1 min and 95°C for 10 min, followed by 40 cycles of denaturation at 95°C for 15 sec and annealing/extension at 60°C for 1 min. The primer sequences used for the PCR were designed by the Primer-3 program and listed as follows: αSyn, 5′-AATGCCTTCTGAGGAAGGGTA-3′ (forward), 5′-CAAATTTTGTAATCCAGAGGTTGA-3′ (backward); 18S rRNA, 5′-CAGCCACCCGAGATTGAGCA-3′ (forward), 5′-TAGTAGCGACGGGCGGTGTG-3′ (backward); GADPH, 5′-GGTGGTCTCCTCTGACTTCAACA-3′ (forward), 5′-GTTGCTGTAGCCAAATTCGTTGT-3′ (backward); HMBS, 5′-ACGGCTCAGATAGCATACAAGAG-3′ (forward), 5′-GTTACGAGCAGTGATGCCTACC-3′ (backward).
Agarose-gel electrophoresis of PCR products and melting-curve analysis were performed to verify the specificity and identity of the PCR products. The mRNA level of αSyn was normalized to that of the reference genes (18S rRNA, GAPDH, or HMBS) and are presented as fold changes relative to control using the 2–ΔΔ Ct method [45]. The geometric mean values of fold changes were calculated based on three selected reference genes. Data are expressed as means±standard error (SEM). Statistical differences between groups are evaluated by one-way analysis of variance (ANOVA) followed by post hoc Dunnett’s test. Statistical significance is considered when the p-value is <0.05.
Immunofluorescence staining analysis
HEK293 cells (2×105 cells/well) were grown on 12 mm glass coverslips in 24-well plates overnight and then transfected with the plasmids (0.5 μg) encoding FLAG-tagged proteins, using TransIT-X2 transfection reagents (#MIR6000, Mirus). At 48 h post-transfection, cells were washed once with PBS, fixed with 4% formaldehyde in PBS for 10 min, and then permeabilized with 0.3% Triton X-100 in PBS for 10 min. Cells were next incubated with 4% FBS in PBS for 30 min to block nonspecific binding of antibodies and then stained with an Alexa-Fluor-594-conjugated anti-FLAG rat monoclonal antibody (1 : 500; #637313, BioLegend) for 1 h, followed by washing with PBS three time for 5 min each. Cells were fixed again with 4% formaldehyde for 10 min and then washed once with distilled water. The coverslips were then placed onto the microscope slide mounted with drops of antifade mounting medium (#H-1200, Vector Laboratories), which contains 4′-6-diamidino-2-phenylindole (DAPI, 1.5 μg/ml) for localizing the nucleus. Cells were then examined at 40×magnification by a fluorescence microscope equipped with a mercury lamp. The fluorescence images were captured by a Nikon digital camera and merged together as two-color TIFF images using the ImageJ software. In the cells transfected with the plasmids (0.5 μg) encoding V5-tagged αSyn, together with the plasmids (0.5 μg) encoding different single-chain antibodies against αSyn, a fluorescein isothiocyanate (FITC)-conjugated anti-V5 mouse monoclonal antibody (1 : 500; #R-963-25, ThermoFisher Scientific) was used to probe αSyn at 48 h post-tranfection, following the staining procedure as described above. Cells were examined at 20× magnification, and the fluorescence integrated density on the images acquired from six microscopic fields with >90% cell confluence was measured using the ImageJ software. To standardize the variables that could affect signal intensity in the immunohistochemistry (IHC), we used the same batch of reagents to perform IHC, where each sample was incubated with antibodies for the same time and treated with the same wash procedure. Also, all immunofluorescence images were taken in the same period by the same microscope/camera system under the same setting of light emission/excitation and exposure time.
Animals and in vivo study
Adult male Sprague-Dawley (SD) rats were purchased (BioLASCO Co., Ltd. Taiwan) at eight weeks of age and housed under environmentally controlled conditions for one week before the initiation of in vivo studies. Rats were anesthetized with chloral hydrate (400 μg/kg, i.p.) and then mounted on a classical stereotaxic frame. A burr hole was drilled in the skull. For investigating the effect of NAC32 on the protein level of overexpressed αSyn, rats were stereotactically injected with AAV-αSyn (1 μ1, 5.4×108 viral genome copy [VGC]/μ1) combined with AAV-NAC32 (1 μ1, 2.7×108 VGC/μ1) or AAV-sc6H4 (1 μ1, 2.7×108 VGC/μ1) into the substantia nigra, according to the following coordinates: anteroposterior (AP): –5.2 mm from the bregma, mediolateral (ML): –2.2 mm from the midline, dorsoventral (DV): –7.0 mm from the dura. For investigating the effect of NAC32 on the protein level of endogenous αSyn, rats were stereotactically injected with AAV-NAC32 (1 μ1, 2.7×108 VGC/μ1) into the substantia nigra or untreated as the control. The injection was performed with a Hamilton Microliter syringe (10 μ1 volume, cemented needle, 22 s gauge, blunt tip) controlled by a microsyringe pump at an infusion rate of 0.2 μ1/min. A few weeks after brain injection, animals were sacrificed, and the brain tissues (substantia nigra, striatum) were collected for western blot analysis. The animal protocol was reviewed and approved by the Animal Research Ethics Board at National Health Research Institutes in Taiwan (Permit Number: NHRI-IACUC-105018-A). While conducting the experiments, we have made the best effort to avoid, minimize, and alleviate stress on animals.
Statistical analysis
All values were expressed as means±standard error (SEM) and analyzed by GraphPad Prism 6.0 statistical software. The difference between two groups was assessed by unpaired, two-tailed Student’s t-test. Comparisons between multiple groups were performed by one-way or two-way analysis of variance (ANOVA) followed by Dunnett’s or Sidak’s post hoc test, respectively, as appropriate. Differences were considered significant when the p-value is <0.05.
RESULTS
Generation and characterization of genetic constructs of αSyn and anti-αSyn intrabodies
The human αSyn gene was genetically fused with a V5-tag coding sequence at the 5′-end and cloned into an adeno-associated virus shuttle plasmid, resulting in the plasmid pAAV-αSyn (Fig. 1A). To examine the expression of αSyn, we transfected HEK293 cells with pAAV-αSyn. At 48 h post-transfection, the whole-cell lysates of transfected cells were fractionated and examined by western blot analysis. A single protein band with a molecular weight similar to the predicted size of the V5-tagged αSyn (16.2 kDa) was detected by an HRP-conjugated anti-V5 antibody in the supernatant fraction, but not in the pellet fraction, of the whole-cell lysates (Fig. 1B). In addition, the transfected cells were subjected to immunofluorescence staining using an FITC-conjugated antibody against the V5 tag. The expression of αSyn was evenly distributed in the cytoplasmic compartment (Fig. 1C). These data suggest that the V5-tagged αSyn is expressed as a soluble monomeric protein in the cytoplasm.
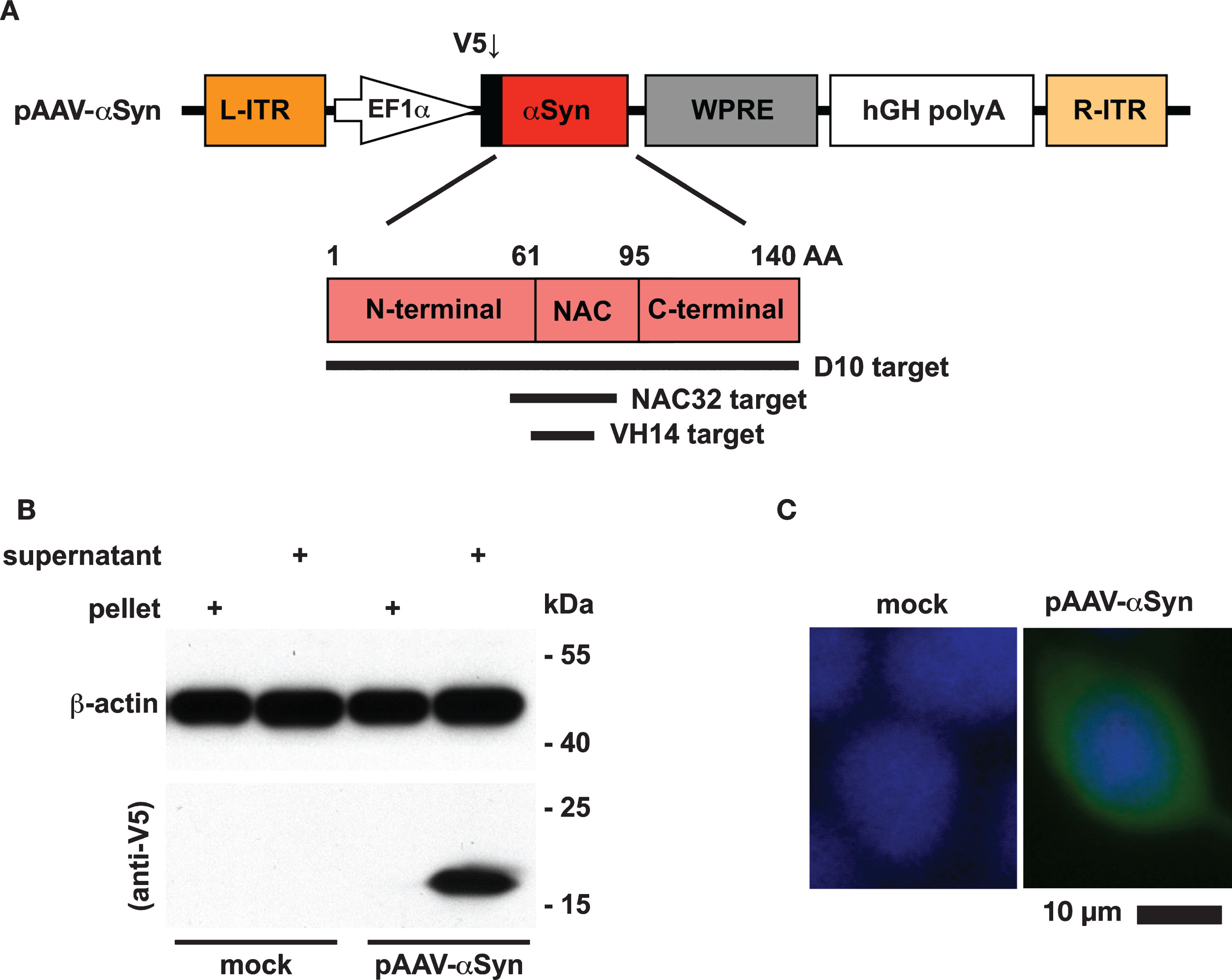
Protein expression of αSyn. A) The human αSyn gene was genetically fused with a V5 tag at the 5′-end and driven by an EF1α promoter in the adeno-associated virus (AAV) shuttle plasmid pAAV-αSyn. The αSyn protein (140 amino acids) consists of three distinct regions: N-terminal region (1–60 residues), non-amyloid-beta component (61–95 residues), and C-terminal region (96–140 residues). The target regions recognized by three distinct intrabodies (D10, NAC32, VH14) are highlighted by black lines. B) Western blot analysis. HEK293 cells were transfected with or without pAAV-αSyn. At 48 h post-transfection, the supernatant and pellet partitions of whole-cell lysates were subjected to western blot analysis to probe the V5 tag and β-actin with specific antibodies. The molecular weight (kDa) and migration location of protein markers were indicated. C) Immunofluorescence staining analysis. The expression of αSyn in HEK293 cells transfected with pAAV-αSyn was detected by an FITC-conjugated antibody (green) against the V5 tag. Nuclei were labeled with DAPI (blue). Mock: no transfection. Scale bar = 10 μm.
The genes of three intrabodies (NAC32, D10, VH14) against different regions of human αSyn and an intrabody (sc6H4) against methamphetamine, used as a control, were genetically fused with a FLAG-tag coding sequence at the 5′-end and a 6×His tag coding sequence at the 3′-end. Each of these four gene fragments was cloned into an adeno-associated virus shuttle plasmid to generate plasmids pAAV-NAC32, pAAV-D10, pAAV-VH14, and pAAV-sc6H4, respectively (Fig. 2A). To evaluate the expression of these intrabodies, we transfected HEK293 cells with these four plasmids separately and examined the whole cell lysates by western blot analysis with an HRP-conjugated anti-FLAG antibody. The whole cell lysates were partitioned into the supernatant fractions (containing soluble proteins) and pellet fractions (containing insoluble proteins) by centrifugation. A single protein band migrating to the position near the predicted molecular weight of each FLAG-tagged intrabody (NAC32, 28.7 kDa; D10, 26.7 kDa; VH14, 17.9 kDa; sc6H4, 27.8 kDa) was mostly identified in the pellet fraction of transfected cell lysates. D10 and sc6H4 intrabodies were also detected in the supernatant fraction but with a much lower intensity than that in the pellet fraction; for the cell lysates of NAC32 and VH14, no visible protein band was detected in the supernatant fraction. (Fig. 2B). We also examined the transfected cells by immunofluorescence staining using an Alexa-Fluor-594-conjugated antibody against the FLAG tag. These four intrabodies were predominantly found in the perinuclear area with punctate staining patterns (Fig. 2C). These results suggest that the FLAG-tagged anti-αSyn intrabodies are expressed as a monomeric protein in the cytoplasmic compartment with low solubility.
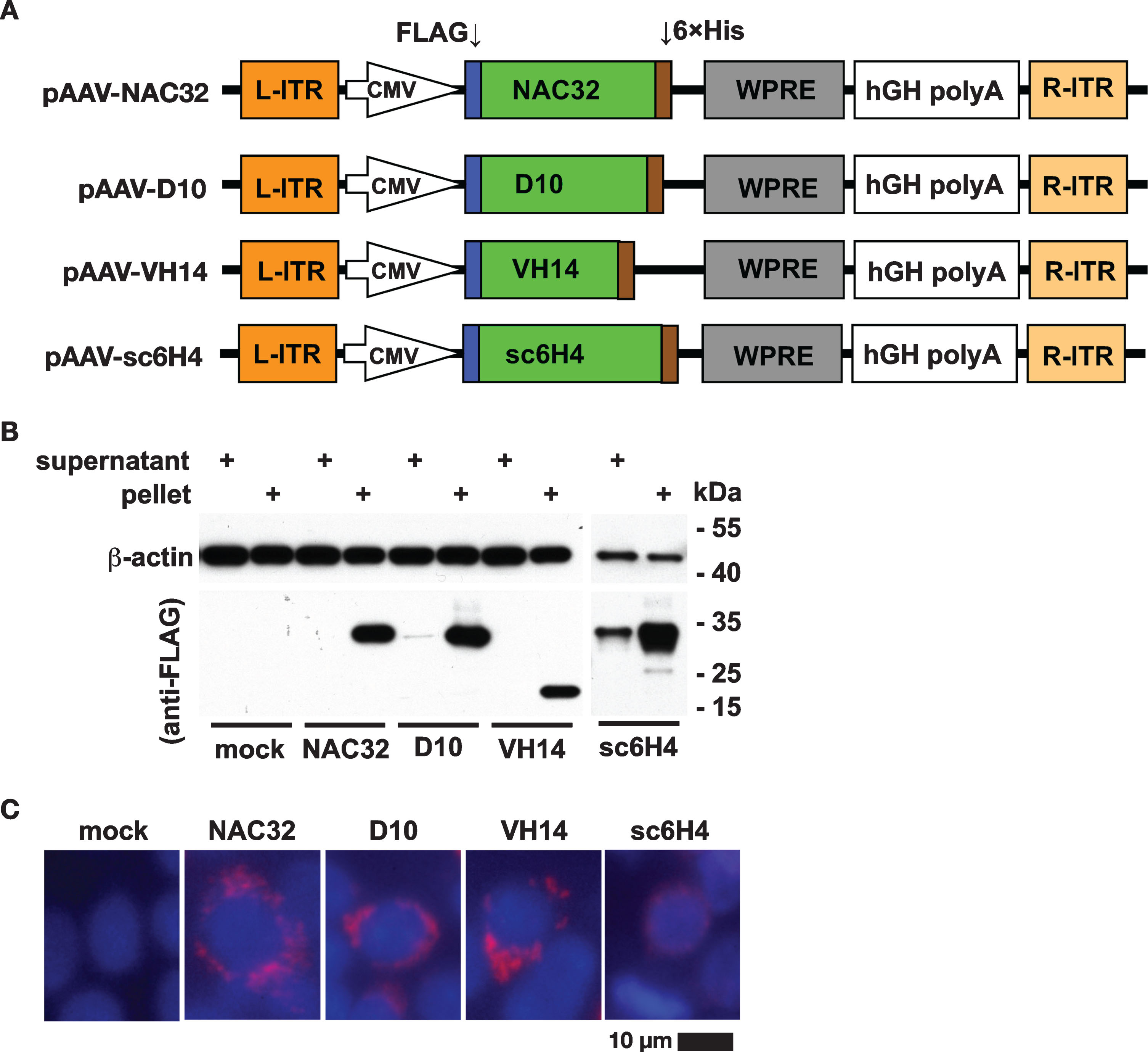
Protein expression of αSyn-specific intrabodies. A) The genes of different intrabodies against αSyn (NAC32, D10, or VH14) and methamphetamine (sc6H4) were genetically fused with a FLAG tag at the 5′-end and a 6× His tag at the 3′-end. The expression of these intrabody genes was driven by a CMV promoter in the adeno-associated virus (AAV) shuttle plasmids (pAAV-NAC32, pAAV-D10, pAAV-VH14, and pAAV-sc6H4). B) Western blot analysis. HEK293 cells were transfected with these four shuttle plasmids. At 48 h post-transfection, the supernatant and pellet partitions of whole-cell lysates were analyzed by western blot using specific antibodies against the FLAG tag and β-actin. All four intrabodies were mainly detected in the pellet (insoluble) partition of cell lysates but rarely detected in the supernatant (soluble) partition of cell lysates, indicating that these four intrabodies were highly insoluble. The molecular weight (kDa) and migration location of protein markers were indicated. C) Immunocytochemical analysis. The expression of each intrabody in transfected HEK293 cells was detected by an Alexa-Fluor-594-conjugated antibody (red) against FLAG tag. Nuclei were labeled with DAPI (blue). Mock: no transfection. Scale bar = 10 μm.
NAC32 intrabody reduced αSyn protein levels in cultured cell lines
To determine the effects of αSyn-specific intrabodies on αSyn protein levels, we transfected HEK293 cells with pAAV-αSyn combined with a plasmid encoding an intrabody (NAC32, D10, or VH14) against αSyn or against methamphetamine (sc6H4) as control. The whole-cell lysates were subjected to western blot analysis at 48 h post-transfection. Compared to the control group, αSyn levels were significantly reduced by 90% in the cells co-transfected with NAC32 (p = 0.0008), reduced by 47% in the cells co-transfected with D10 (p = 0.054, borderline significance), and not apparently affected in the cells co-transfected with VH14 (F3,12 = 11.5; one-way ANOVA, Dunnett’s multiple comparison tests) (Fig. 3A, 3B). To confirm that NAC32 intrabody decreases αSyn protein levels, we examined the transfected cells by immunofluorescence staining, where αSyn was probed by an FITC-conjugated antibody. The total integrated intensity of detected fluorescence in the cells co-transfected with NAC32 (3.0±0.08×108; p < 0.0001), D10 (3.6±0.08×108; p < 0.0001), or VH14 (4.0±0.14×108; p = 0.0134) was significantly lower than that in the cells co-transfected with sc6H4 (4.4±0.04×108 pixels; F3,20 = 39.4, one-way ANOVA, Dunnett’s multiple comparison test) (Fig. 3C, 3D). Among the examined anti-αSyn intrabodies, NAC32 was thus most effective in reducing αSyn protein levels in HEK293 cells, and, therefore, we further evaluated whether NAC32 exhibits similar effects in other cell types by western blot analysis. NAC32 co-transfection also led to a significant reduction of αSyn by 92% in the human neuroblastoma cell line SH-SY5Y (p < 0.0001; Student’s t test) (Fig. 3E, 3F). These results suggest that NAC32 intrabody profoundly reduces the protein levels of αSyn when co-expressed with αSyn in cultured cells.
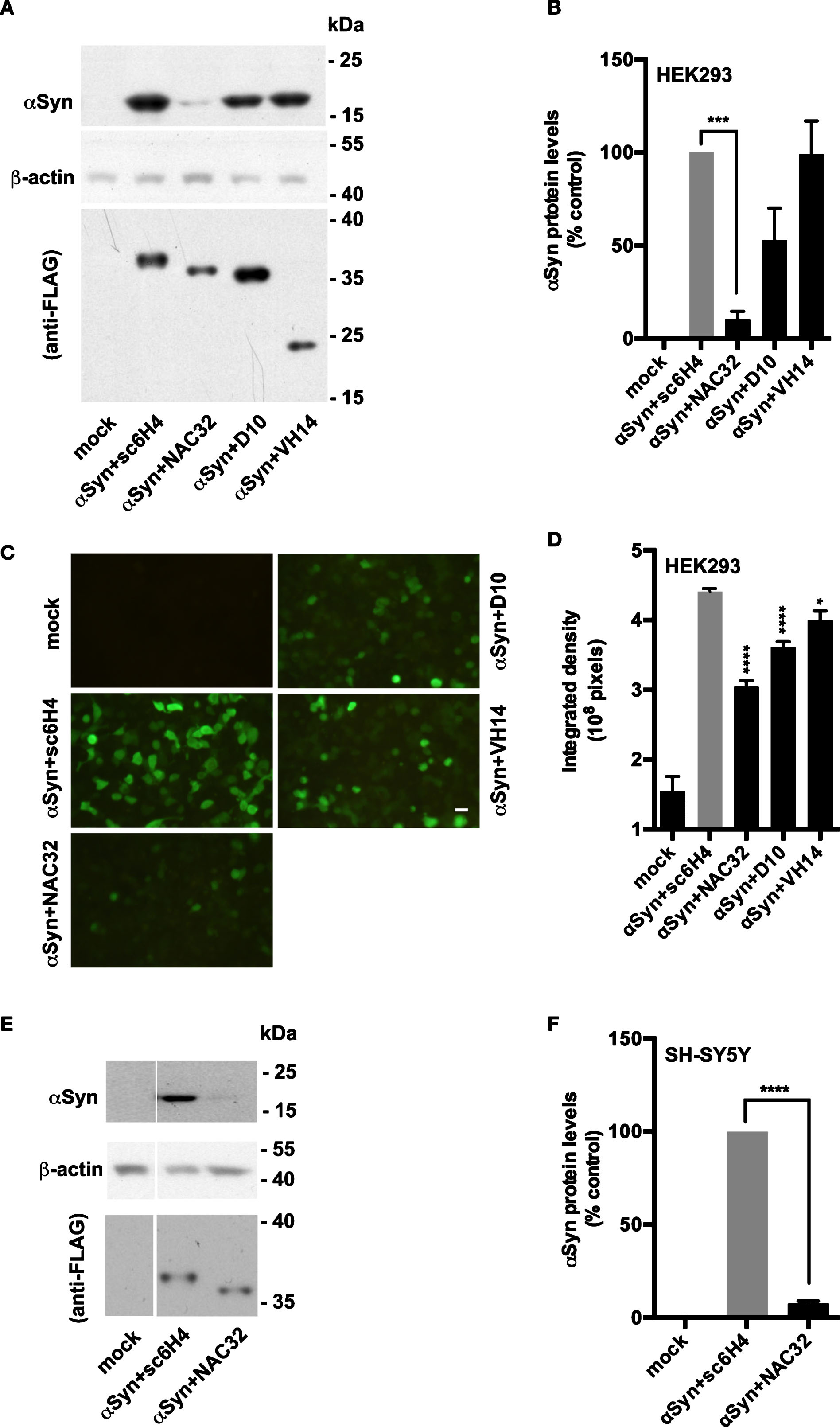
The αSyn-specific intrabody NAC32 decreases αSyn protein levels in cell lines. A) Western blot analysis. HEK293 cells were transfected with pAAV-αSyn combined with plasmids encoding distinct intrabodies binding to αSyn (NAC32, D10, or VH14) or methamphetamine (sc6H4; as control). At 48 h post-transfection, whole-cell lysates were examined by western blot analysis using antibodies against the V5 tag (to probe αSyn), FLAG tag (to probe intrabodies), and β-actin. The molecular weight (kDa) and migration location of protein markers are indicated. A representative result from four independent experiments is shown. B) The expression levels of probed proteins in HEK293 cells were measured by densitometric analysis. αSyn levels were normalized to β-actin levels, and the relative optical density values were presented as percentages of control. Data were expressed as mean values±SEM. C) Immunofluorescence staining analysis. HEK293 cells were transfected with pAAV-αSyn combined with pAAV-sc6H4, pAAV-NAC32, pAAV-D10, or pAAV-VH14, or not transfected (mock). At 48 h post-transfection, cells were subjected to immunofluorescence staining analysis using an FITC-conjugated antibody against the V5 tag to detect αSyn. A set of representative fluorescence images is shown. Scale bar = 20 μm. D) The raw integrated density of fluorescence was measured from images taken from six microscopic fields with >90% cell confluence. Data were expressed as mean values±SEM. E) Western blot analysis. Human neuroblastoma SH-SY5Y cells were transfected with pAAV-αSyn combined with pAAV-sc6H4 or pAAV-NAC32. The whole-cell lysates were examined by western blot analysis as described above. F) The relative protein levels of αSyn in SH-SY5Y cells were measured by densitometric analysis, and the data were presented as described above. Mock: no transfection. Significant differences from the control group are indicated (*p < 0.05; ***p < 0.001, ****p < 0.0001; one-way ANOVA: for B, D; Student’s t-test: for F).
NAC32 intrabody did not alter αSyn mRNA levels
To examine whether NAC32 intrabody-mediated reduction of αSyn protein is caused by the downregulation of αSyn mRNA levels, we transfected HEK293 cells with pAAV-αSyn combined with pAAV-NAC32, pAAV-D10, pAAV-VH14, or pAAV-sc6H4 as control. At 48 h post-transfection, total RNA was extracted for qRT-PCR analysis to quantify αSyn mRNA levels which were normalized to three reference genes (18 S rRNA, GAPDH, and HMBS) and presented as fold changes relative to control using the 2–ΔΔCt method. No αSyn mRNA was detected in the group receiving no transfection. Compared with the control group cotransfected with pAAV-sc6H4 (1.003±0.037), the group cotransfected with pAAV-NAC32 (1.078±0.085, p = 0.5806), pAAV-D10 (1.035±0.022, p = 0.9343), or pAAV-VH14 (0.893±0.021, p = 0.3008) showed no significant difference of αSyn mRNA levels (F3,12 = 2.637; one-way ANOVA, Dunnett’s multiple comparison test; Fig. 4). These data suggest that NAC32 intrabody does not alter αSyn mRNA expression.
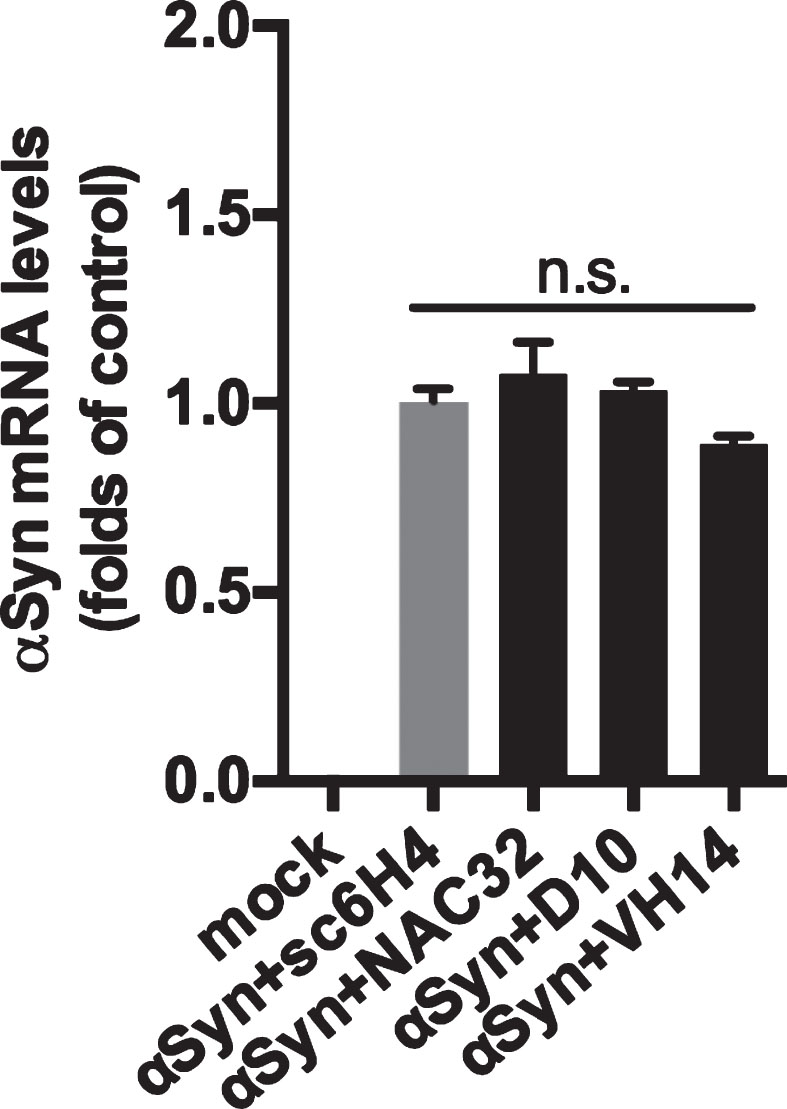
NAC32 intrabody does not reduce αSyn mRNA levels. HEK293 cells were transfected with pAAV-αSyn combined with pAAV-NAC32, pAAV-D10, pAAV-VH14, or pAAV-sc6H4 (as control), or not transfected (mock). At 48 h post-transfection, total RNA was extracted and subjected to qPCR analysis to quantify αSyn mRNA levels, which were normalized to three selected reference genes and presented as folds of control (αSyn + sc6H4). Data were expressed as mean values±SEM. n.s.: not significant (p = 0.0974; one-way ANOVA).
NAC32 intrabody-mediated reduction of αSyn protein levels is proteasome independent
To investigate whether NAC32 intrabody-induced downregulation of αSyn protein is through proteasome-mediated degradation, we transfected HEK293 cells with pAAV-αSyn combined with pAAV-NAC32 or pAAV-sc6H4 as control. At 24 h post-transfection, the medium was supplemented with or without the proteasome inhibitor MG132. After the addition of MG132 for 8 h, cells were harvested for western blot analysis to detect αSyn and the intrabodies NAC32 and sc6H4. The expression levels of αSyn were normalized to β-actin levels. In the cells transfected with pAAV-αSyn + pAAV-NAC32, the level of αSyn was reduced in the absence of MG132 (2.55±0.205 vs. 1.49±0.172, p = 0.0228); however, this effect was not inhibited by the treatment with MG132 (2.707±0.273 vs. 0.787±0.254, p = 0.0007; two-way ANOVA, Sidak’s multiple comparison test) (Fig. 5 A,B). In the same experiment, MG132 increased the levels of NAC32 and sc6H4 intrabodies, indicating that MG132 was effective at blocking the processes of proteasome-mediated degradation. These results suggest that NAC32 intrabody-induced reduction of αSyn is not attributed to proteasome-mediated degradation.
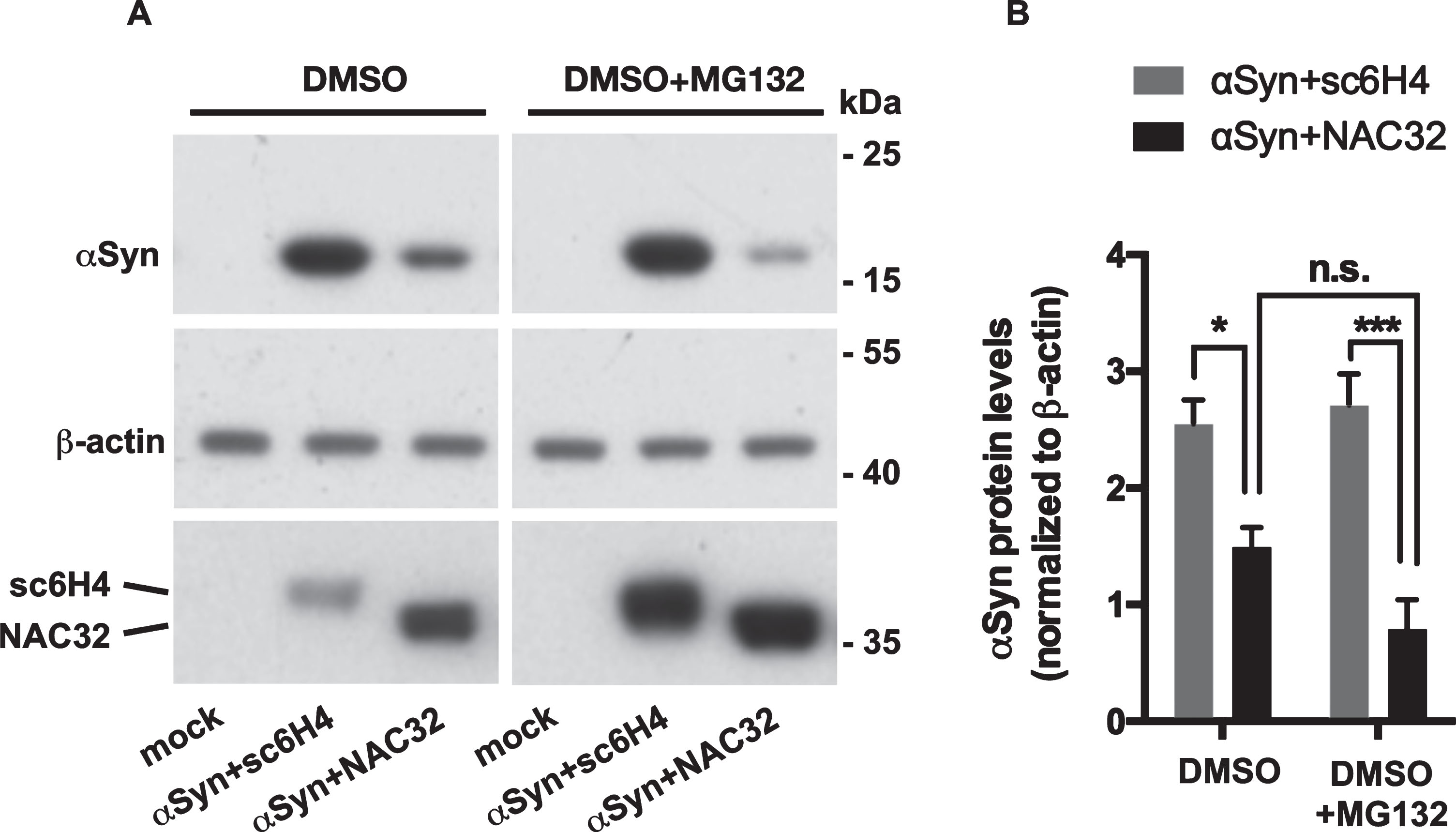
The proteasome inhibitor MG132 does not inhibit NAC32 intrabody-mediated reduction of αSyn protein levels. A) Western blot analysis. HEK293 cells were transfected with pAAV-αSyn combined with pAAV-NAC32 or pAAV-sc6H4 (as control) and maintained in the medium supplemented with 0.2% DMSO or 0.2% DMSO+10 μM MG132. At 8 h post-transfection, cells were harvested and subjected to western blot analysis to detect αSyn (using anti-V5 antibody), intrabodies (NAC32, sc6H4; using anti-FLAG antibody), and β-actin. Mock: not transfected. B) The expression levels of probed proteins were measured by densitometric analysis. αSyn levels were normalized to β-actin levels, and the data were expressed as mean values±SEM. In the cells transfected with pAAV-αSyn + pAAV-NAC32, there was no statistical difference (n.s.) between the treatment with DMSO and DMSO + MG132. Significant differences from the control group are indicated (*p < 0.05, ***p < 0.001; two-way ANOVA).
NAC32 intrabody reduces αSyn protein levels and attenuates the loss of tyrosine hydroxylase immunoreactivity in the rat striatum
We next explored whether NAC32 intrabody reduced αSyn expression in vivo. Given the in vitro data detailed above, we only used NAC32 and sc6H4 intrabody constructs; AAV particles encoding αSyn, NAC32, or sc6H4 were generated and purified by an AAV-specific affinity column. AAV-αSyn (1 μl; 5.4×108 VGC/μl) combined with either AAV-NAC32 (1 μl; 2.7×108 VGC/μl) or AAV-sc6H4 (1 μl; 2.7×108 VGC/μl) was administered into the substantia nigra in adult rats (n = 6; Fig. 6A). On 30 days after AAV injection, substantia nigra and striatal tissues were collected for western blot analysis to detect αSyn, tyrosine hydroxylase (TH), intrabodies (NAC32, sc6H4), and β-actin. The expression levels of detected proteins were measured by densitometric analysis, and αSyn and TH levels were normalized to β-actin levels. NAC32 was detected in the substantia nigra of the rats injected with AAV-αSyn + AAV-NAC32, while sc6H4 was detected in the substantia nigra and striatum of the rats injected with AAV-αSyn + AAV-sc6H4 (Fig. 6B, 6C). The AAV-encoded αSyn levels were significantly lower in the rats injected with AAV-αSyn + AAV-NAC32 than in rats injected with AAV-αSyn + AAV-sc6H4 (substantia nigra: 0.683±0.128 vs. 1.271±0.117, p = 0.0276, Fig. 6B, 6D; striatum: 0.172±0.058 vs. 0.922±0.249, p = 0.0429, Fig. 6C, 6E; unpaired two-tailed Student’s t test). In the striatum, TH levels were significantly higher in the rats injected with AAV-αSyn + AAV-NAC32 than in rats injected with AAV-αSyn + AAV-sc6H4 (1.202±0.112 vs. 0.629±0.040, p = 0.0084, Fig. 6C, 6G; unpaired two-tailed Student’s t-test). No significant change of TH levels was found in the substantia nigra (0.969±0.099 vs. 0.874±0.182, p = 0.6694, Fig. 6B, 6F). These results suggest that AAV-mediated expression of NAC32 intrabody can lead to a reduction of αSyn and attenuate the reduction of TH expression. However, whether NAC32 intrabody can prevent degeneration of dopaminergic terminals in the striatum requires further experiments to examine the survival and/or loss of these neuronal fibers in the brain tissue.
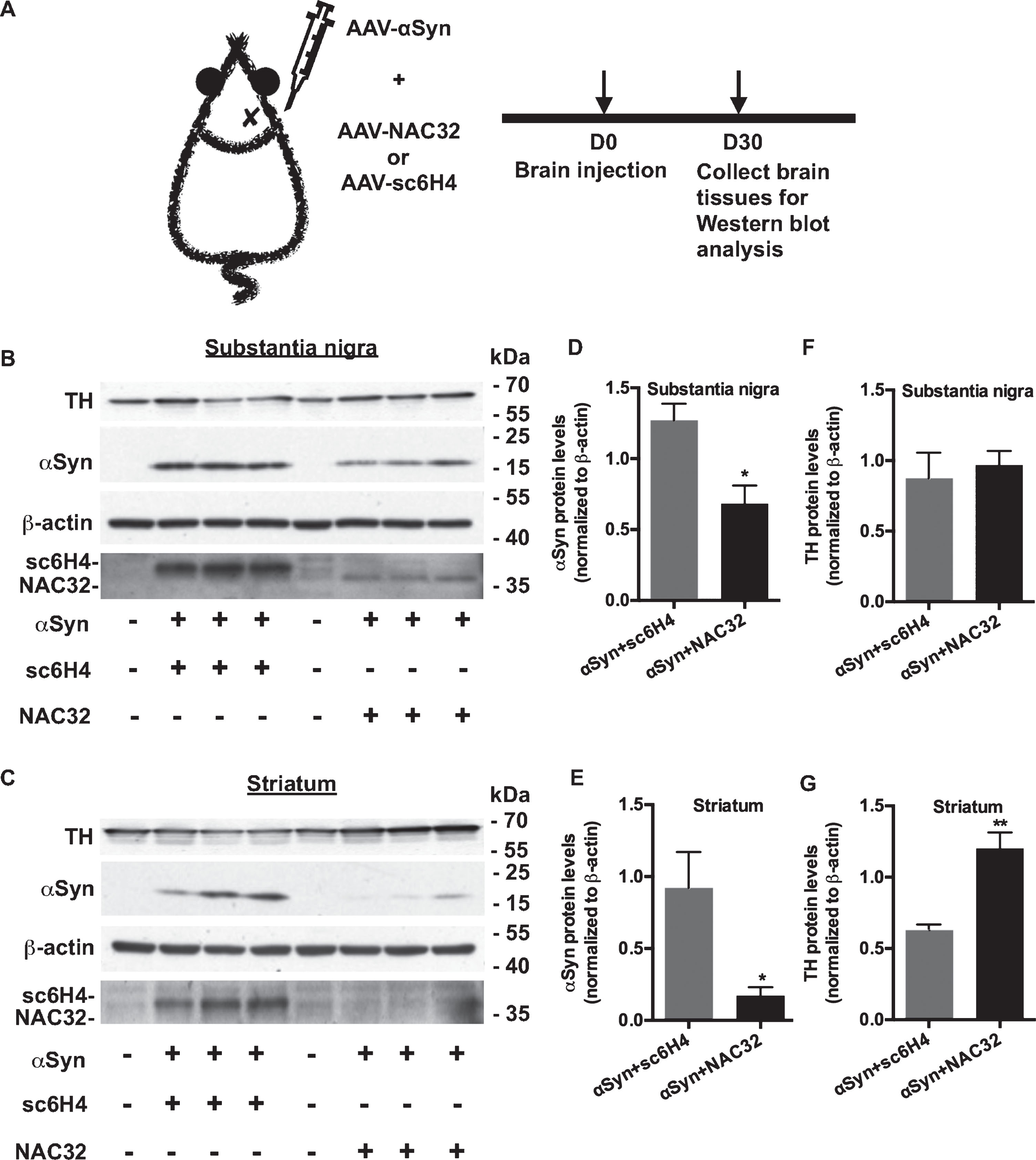
NAC32 intrabody reduces αSyn but not tyrosine hydroxylase protein levels in the rat brain. A) Experiment timeline. Adult rats (n = 6) were stereotactically injected with AAV-αSyn + AAV-sc6H4 (as control) and AAV-αSyn + AAV-NAC32 into the right substantia nigra. B, C) Western blot analysis. On 30 days after injection, substantia nigra and striatal tissues were collected for western blot analysis to detect αSyn (using anti-V5 antibody), tyrosine hydroxylase (TH), intrabodies (NAC32, sc6H4; using anti-FLAG antibody), and β-actin. D–G) The expression levels of probed proteins were measured by densitometric analysis. αSyn and TH levels were normalized to β-actin levels, and the data were plotted as mean values±SEM. Significant differences were indicated (*p < 0.05, **p < 0.01; unpaired, two-tailed Student’s t-test versus control).
NAC32 intrabody downregulates endogenous αSyn protein levels in the rat brain
We further investigated whether NAC32 can reduce the expression of endogenous αSyn in vivo. Adult rats were unilaterally injected with AAV-NAC32 (1 μl; 2.7×108 VGC/μl) into the substantia nigra (n = 6) or untreated as the control (n = 8; Fig. 7A). On 25 days after AAV injection, animals were sacrificed, and substantia nigra tissues were collected for western blot analysis to detect endogenous αSyn, and β-actin. The expression levels of detected proteins were measured by densitometric analysis, and the endogenous αSyn levels were normalized to β-actin levels. The endogenous αSyn level was significantly lower in the rats injected with AAV-NAC32 than that in the untreated rats (0.772±0.128 vs. 1.328±0.977, p = 0.0042, Fig. 7B, 7C; unpaired two-tailed Student’s t-test). These results suggest that AAV-mediated gene delivery of NAC32 intrabody can reduce the expression of endogenous αSyn in vivo.
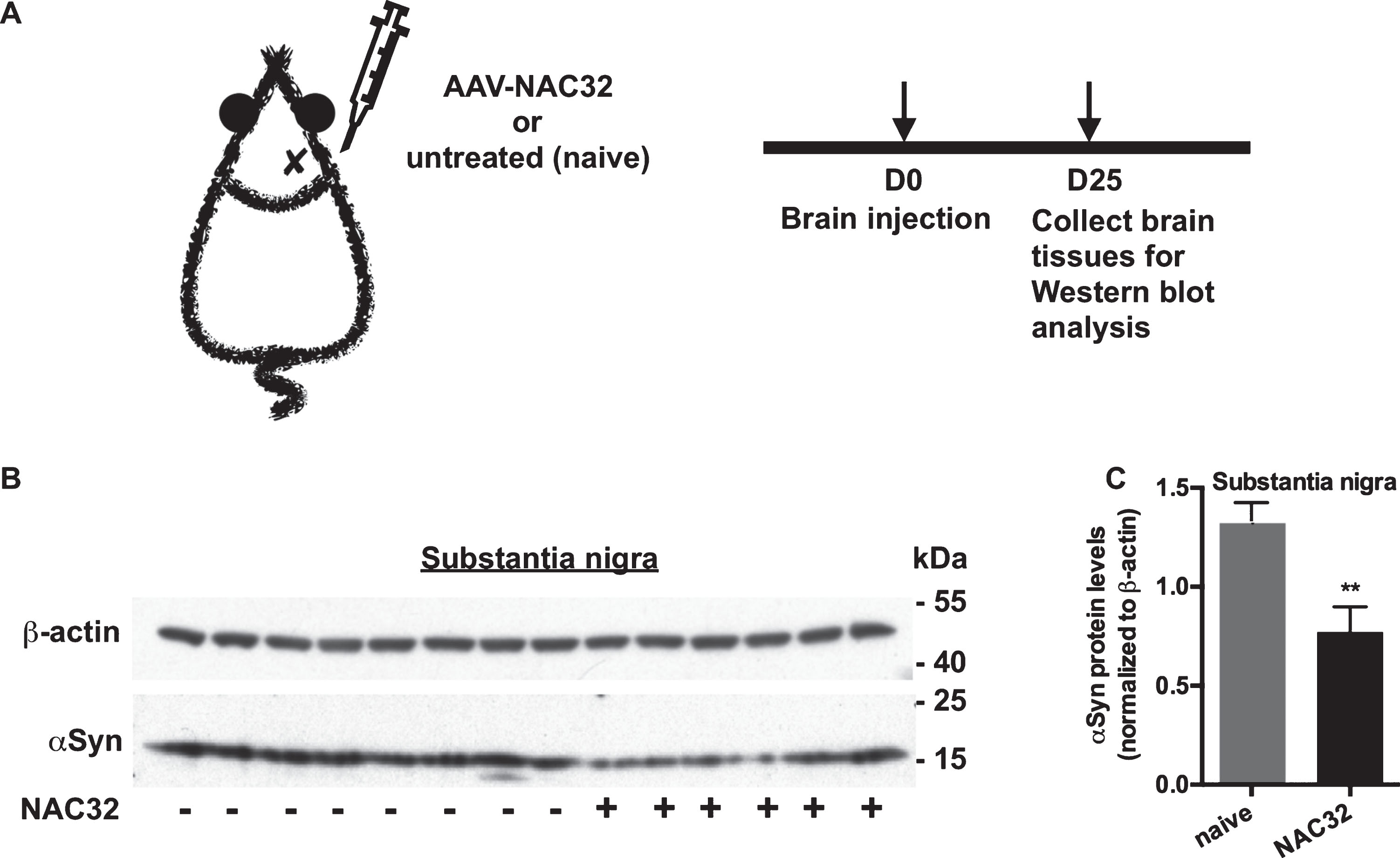
NAC32 intrabody reduces endogenous αSyn protein levels in the rat brain. A) Experiment timeline. Adult rats (n = 14) were stereotactically injected with AAV-NAC32 into the right substantia nigra or untreated as the control (naive). B) Western blot analysis. On 25 days after injection, substantia nigra tissues were collected for western blot analysis to detect endogenous αSyn, and β-actin. C) The expression levels of probed proteins were measured by densitometric analysis. Endogenous αSyn levels were normalized to β-actin levels, and the data were plotted as mean values±SEM. Significant difference was indicated (**p < 0.01; unpaired, two-tailed Student’s t-test versus control).
DISCUSSION
The objective of this study was to investigate whether αSyn-specific intrabodies reduce human αSyn protein levels in vitro and in vivo. Our data showed that, among three intrabodies examined in cell cultures, NAC32 intrabody was demonstrated to selectively reduce αSyn protein levels in both HEK293 and SH-SY5Y cell lines. Moreover, it is unlikely that this protein downregulation was a result of mRNA reduction or proteasome-mediated degradation, and the underlying mechanisms remain elusive. Nevertheless, this reduction was also found in an in vivo model, where intracranial injection of the AAV encoding NAC32 intrabody significantly reduced the protein levels of AAV-encoded and endogenous αSyn in the rat brain. To our knowledge, this is the first report demonstrating that NAC32 intrabody can reduce αSyn protein levels in human cell lines and the rat brain. Although further investigations are needed, these results suggest that AAV-mediated expression of αSyn-specific intrabodies in the brain is a potential treatment for the diseases associated with αSyn accumulation.
Reduction of intracellular protein accumulation can be achieved by RNA interference-mediated post-transcriptional gene-silencing and/or intrabody-directed post-translational modification of target proteins [46]. Although the RNA interference approach has been successfully used to downregulate αSyn and rescue αSyn-mediated behavioral deficits in rodents, chronic expression of short-hairpin RNA targeting αSyn mRNA also leads to dopamine neuron loss and substantia nigra inflammation [47, 48]. In addition, the RNA interference approach poses inherent disadvantages: off-targeting effects caused by sequence mismatch and non-specific gene regulation arising primarily from interferon stimulation triggered by short double-stranded RNA [49–51]. Therefore, due to safety concerns for future translation, the application of RNA interference to reduce αSyn accumulation for the treatment of Parkinson’s disease remains challenging. Alternatively, intrabodies can be pre-selected for high specificity for the disease proteins and delivered as genes, while their expression does not generate short double-stranded RNA. It has been reported that an intrabody, which preferentially recognizes mutant huntingtin protein, can facilitate the clearance of cytoplasmic huntingtin through increasing ubiquitination-associated degradation in neuronal processes of Huntington disease mouse brain without causing intrabody-associated neuron damage [52]. However, there is still a lack of evidence demonstrating that intrabodies targeting αSyn do not cause damage to the brain.
In the current study, we have demonstrated that simultaneous co-expression of αSyn and the αSyn-specific intrabody NAC32 by plasmid transfection or AAV transduction can lead to the downregulation of αSyn protein levels in the cultured cells and rat brain. This effect might not be caused by artificial impacts derived from the delivery of exogenous genes for αSyn expression since AAV-mediated gene delivery of NAC32 also reduced the expression of endogenous αSyn in the rat brain. In addition, NAC32-mediated reduction of αSyn seems to be independent of proteasome-mediated degradation and does not act at the post-transcriptional level; the proteasome inhibitor does not prevent the intrabody-mediated reduction of αSyn and expression of NAC32 does not change αSyn mRNA levels. In previous studies, it has been demonstrated that αSyn-specific antibodies produced by immunization in mice might enter the neurons through binding to surface receptors or membrane-bound αSyn and then promote degradation of intracellular αSyn aggregates in the brain via lysosomal pathways [53]. Recently, it was reported that an anti-Tau intrabody, engineered to fused with a mutant ubiquitin favoring polyubiquitination at K63, was able to target intracellular Tau proteins for lysosomal degradation [54]. Further studies are warranted to examine whether the lysosomal pathway is involved in the degradation of αSyn, following NAC32 intrabody treatment. Although the mechanism of NAC32-associated reduction of αSyn remains elusive, it is worth further investigating whether NAC32 can reverse intracellular αSyn accumulation without causing detrimental side effects in the rodent brain, which has already over-expressed αSyn.
Although intrabodies are ideal reagents for modulation of intracellular disease proteins, their application has been hampered because the majority of intrabodies are poorly soluble in the cytoplasm. We observed that the αSyn-specific intrabody VH14 was highly insoluble in the cytoplasm when expressed in mammalian cells, consistent with a previous report [55]. The low solubility of VH14 may alter the functional structure of this intrabody and prevent binding to and degredation of αSyn. Similar to VH14, D10 is also poorly soluble in the cytoplasm. However, D10 significantly inhibited the formation of oligomeric αSyn and ameliorated the cell-adhesion defect in HEK293 cells over-expressing αSyn [42]. When transiently co-transfecting αSyn with D10, we found a trend (p = 0.054) for αSyn reduction by western blot analysis and a significant αSyn reduction (p < 0.0001) by immunofluorescence staining analysis, suggesting that the latter is more sensitive in detecting intrabody-mediated αSyn reduction. Further investigation is required to validate D10’s capacity for regulating αSyn levels.
Several attempts have been reported to improve the solubility of intrabodies [55, 56]. For example, VH14 fused with the highly charged proteasomal retargeting sequence (enriched in residues P, E, S, and T or PEST) improved solubility of VH14, promoted proteasome-mediated degradation of αSyn, and decreased αSyn toxicity in a rat neural progenitor cell line [55, 57]; also, it alleviated pathology and motor functional decline in an AAV-mediated αSyn over expression rat PD model [58]. Interestingly, in current study, we demonstrated that NAC32 is also not soluble in the cytoplasm but efficiently down regulates αSyn. Our data support that the low solubility of NAC32 did not alter its function. Indeed, NAC32 has been demonstrated to bind to αSyn intracellularly as seen by the increase in translocation of αSyn/NAC32-NLS (nuclear-localization-signal peptide) from cytoplasm to nucleus [44]. Furthermore, when stably expressed in the rat neural progenitor cell line ST14A, NAC32 significantly reduced intracellular aggregation and toxicity of A53T mutant αSyn [44]. Here, we demonstrated NAC32’s functionality on αSyn down regulation, supporting that NAC32 is a promising therapeutic candidate targeting αSyn for PD. It would be of great interest to investigate whether adding the PEST motif further enhances NAC32’s functionality on downregulation of αSyn.
The expression of intrabodies against αSyn in dopaminergic neurons for the treatment of PD relies on an in vivo gene transfer approach. Viral vectors, such as AAV and lentivirus, are frequently used to achieve effective gene transfer. AAV is a safe and ideal viral vector for gene therapy of neurological diseases [59, 60]. It can transduce various cell types in the central nervous system and achieve long-term stable gene expression in slowly dividing and non-dividing cells, such as neurons, without eliciting deleterious inflammation in the brain [60, 61]. Furthermore, unlike lentivirus vectors, AAV vectors do not integrate genetic materials into the chromosomes of infected cells, which greatly reduces the risk of oncogene activation [62]. In addition, wild-type AAV has low immunogenicity and has not been linked to any human diseases [63, 64]. In the current study, we observed a sustained expression of NAC32 and sc6H4 intrabodies in rat brain after gene delivery through stereotactic injection of serotype-1 AAV vectors, supporting the feasibility of AAV-mediated gene delivery in the central nervous system. Importantly, compared with sc6H4 (methamphetamine specific), NAC32 (αSyn specific) significantly reduced AAV-mediated expression of αSyn in the substantia nigra and striatum areas and increased tyrosine hydroxylase immunoreactivity in the striatum, demonstrating the efficacy of AAV-NAC32 on the regulation of αSyn levels and the possible protection against αSyn-mediated dopaminergic terminal degeneration.
In contrast to the AAV-mediated overexpression of αSyn, exogenous αSyn pre-formed fibrils (PFFs) can trigger more aggregation of endogenous αSyn when introduced in vitro and in vivo. The PFF model of PD shows a more prolonged time course of neurodegeneration and dopamine dysfunction in the PD-relevant brain regions, which is more similar to the clinical condition [66, 67]. Besides the AAV-based PD model used here, the αSyn PFF model could be valuable for investigating the therapeutic potential of AAV-NAC32. In the present study, we have demonstrated that AAV-mediated gene delivery of NAC32 can lead to a significant downregulation of endogenous αSyn in the rat brain. Therefore, stereotactically injection of AAV-NAC32 into the substantia nigra is likely to reduce aggregation of endogenous αSyn in the αSyn PFF model. However, whether AAV-mediated gene transfer of intrabodies against αSyn accumulation can provide therapeutic benefits for PD patients remains to be determined. Nevertheless, intrabody-based gene studies, detailed here, have shed some light on a therapeutic strategy for neurodegenerative conditions associated with abnormal protein aggregation.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
Footnotes
ACKNOWLEDGMENTS
This work was supported in part by grants from Ministry of Science and Technology Taiwan (MOST 106-2314-B-030-005, MOST 107-2314-B-030-009, MOST 108-2314-B-030-007), the College of Science and Engineering at Fu-Jen Catholic University (A0206004), and the National Health Research Institutes Taiwan. We are grateful to Dr. Enamul Ahsan (Anglia Ruskin University, UK) for editorial assistance on this manuscript.