
Abstract
Background:
Sporadic Parkinson’s disease (PD) patients have lower α-galactosidase A (α-GAL A) enzymatic activity and Fabry disease (FD) patients potentially carry an increased risk of PD.
Objective:
Determination of PD prevalence in FD and clinical, biochemical and vascular neuroimaging description of FD pedigrees with concomitant PD.
Methods:
Clinical screening for PD in 229 FD patients belonging to 31 families, harbouring GLA gene mutation p.F113L, and subsequent pedigree analysis. Gender-stratified comparison of FD+/PD+ patients with their family members with FD but without PD (FD+/PD–) regarding Mainz scores, plasma & leukocytes α-GAL A enzymatic activity, urinary Gb3 and plasma Lyso-Gb3, vascular brain neuroimaging.
Results:
Prevalence of PD in FD was 1.3% (3/229) (3% in patients aged ≥50 years). Three FD patients, one female (73 years old) (P1) and two males (60 and 65 years old) (P2 and P3), three different pedigrees, presented akinetic-rigid PD, with weak response to levodopa (16% – 36%), and dopaminergic deficiency on 18F-DOPA PET. No pathogenic mutations were found in a PD gene panel. FD+/PD+ patients had worse clinical severity of FD (above upper 75% IQR in Mainz scores), and cortico-subcortical white matter/small vessel lesions. P3 patient was under enzyme therapy, started 1 year before PD diagnosis. P2-P3 patients had higher leucocyte α-GAL A activity (2,2-3 vs.1,0 (median)(nmol/h/mg)).
Conclusion:
We have shown a high prevalence of PD in a late-onset phenotype of FD, presenting high cerebrovascular burden and weak response to levodopa. Further studies will untangle how much of this PD phenotype is due to Gb3 deposition versus cerebrovascular lesions in the nigro-striatal network.
INTRODUCTION
Anderson-Fabry Disease (FD) is a rare X-linked lysosomal storage disorder caused by deficient activity of the enzyme α-galactosidase A (α-GAL A), resulting in progressive and diffuse lysosomal accumulation of neutral glycosphingolipids, especially globotriaosylceramide (Gb3) in vascular endothelium, kidneys, heart, brain, skin, cornea and other tissues leading to multiorgan damage [1]. Transient ischemic attacks (TIAs), strokes [2] and brain white matter lesions (WML) [3] are common central nervous system (CNS) manifestations. In recent years there has been growing body of evidence supporting a link between the pathogenesis of PD and lysosomal dysfunction, namely in Gaucher disease (GD) and FD.
The potential molecular link was primarily driven by the description of small series of atypical Parkinsonism in Gaucher type I [4] and in “non-neuronopathic” GD [5, 6], and later on by the description of increased risk of developing PD in asymptomatic carriers of GBA gene mutations and relatives of GD patients [4, 7]. From a biological standpoint, the autophagic-lysosomal system has been described to play a major role in α-synuclein aggregation and, in that manner, the loss-of-function of the lysosomal glucocerebrosidase (GCase) affects the processing and clearance of α-synuclein, acquainting for the connection between GD and PD [8].
Parnetti et al., when analysing the cerebrospinal fluid of PD patients (GBA mutation carriers versus noncarriers) and healthy subjects, found that PD patients had not only lower GCase (especially lower in GBA mutation carriers), but also lower cathepsin D and β – hexosaminidase [9]. Alcalay et al. [10], besides GCase, measured α-GAL A, acid sphingomyelinase (deficient in Niemann–Pick disease types A and B), acid α-glucosidase (deficient in Pompe disease) and galactosylceramidase (deficient in Krabbe disease) in dried blood spots of PD patients (n = 648) and healthy subjects (n = 317). The authors adjusted for age, gender, and GBA and LRRK2 G2019S mutation status and showed that PD patients (sporadic plus carriers of LRRK2 G2019S and GBA mutations) have lower GCase and α-GAL A enzymatic activity [10]. These results are in agreement with the study of Wu et al., where lower levels of α-GAL A were found in the leukocytes of sporadic PD patients (n = 38), when compared to age- and gender-matched healthy controls (n = 258) (PD: 18.56±1.49; Controls: 22.24±0.60 units) [11].
Following two clinical reports of FD patients that later on developed parkinsonism [12, 13], Wise et al. [14] performed an online survey and family history questionnaire to determine the prevalence of PD in 90 FD patients. The authors included 63 females and 27 males and identified 2 PD patients (2.2%), carrying the p.Y134X mutation and the p.E59V variant in the GLA gene, plus 2 additional PD patients among their patients older than 60 years of age (2/24, 8.3%). Family history was available in 81 families with FD, and 6 (7.4%) had one first degree relative who fit the criteria for a conservative diagnosis of PD. These observations supported the hypothesis that there may be an increased risk of developing PD in individuals with GLA gene mutations.
Herein we present the prevalence of PD in a large cohort of 229 FD patients, and analyse the pedigrees of three FD patients with concomitant PD, with respect to clinical, biochemical and vascular brain neuroimaging markers.
METHODS
This research was approved by the hospital Ethics Committee of the Hospital Senhora da Oliveira, Guimarães, Portugal (Ref. CAc 42/2018). The study was thoroughly explained to all patients, who provided written informed consent. We have included 229 adult patients (≥18 years) from 31 families carrying the GLA gene mutation c.337T>C (p.F113L). The enzymatic activity of α-GAL A in plasma and leukocytes, the plasma and urinary Gb3 and the plasma lyso-Gb3 were measured according to previously described methods [15–17].
FD patients were submitted to a biannual follow-up, including a systematic multidisciplinary clinical assessment with characterization of the multiorgan involvement by FD [18]. Mainz severity score index (MSSI) was estimated in all patients. The neurological evaluation was performed by the same neurologist and was focused on central and peripheral nervous system complications associated with FD (cerebrovascular disease, cognitive and behaviour complications, depression, anxiety, acroparesthesias, neuropathic pain). Mainz score (neurological sub-score) [19] and general neuropsychological evaluation by Montreal Cognitive Assessment (MoCA) [20] were also evaluated. Brain imaging, by magnetic resonance imaging (MRI) or computed tomography in patients with pacemaker, was performed in all patients, and qualitative description of the vascular brain imaging findings (presence and location of white matter/ischemic/haemorrhagic lesions, global cortical atrophy scale) was specifically collected for the current study based on neuroradiology records. Comorbid cardiac and cerebrovascular risk factors (previous transient ischemic attack, previous ischemic/haemorrhagic stroke, cardiac disease, hypertension, dyslipidaemia, diabetes, smoking) were collected based on clinical records. Patients with FD were diagnosed with parkinsonism according to MDS-clinical criteria for PD [21], and grouped as FD+PD+, and performed a brain 18F-DOPA PET. Genetic testing targeting the major known PD genes (ANO3, ATP13A2, ATP1A3, ATP6AP2, C190RF12, CHCHD2, COASY, CSF1R, DCTN1, DNAJC6, EIF4G1, FBXO7, FTL, GBA, GCH1, GNAL, GRN,LRRK2, MAPT, NR4A2, OPA3, PANK2, PARK7, PDE10A, PDGFB, PDGFRB, PINK1, PLA2G6, POLG, PRKN, PRKRA, RAB39B, SGCE, SLC18A2,SLC20A2, SLC30A10, SLC39A14, SLC6A3, SNCA, SNCAIP, SPG11, SPR, SYNJ1, TAF1, TH, THAP1, TOR1A, TUBB4A, TWNK, UCHL1, VCP, VPS13A,VPS13C, VPS35, WDR45 and XPR1) were requested for PD patients. In order to ascertain family history of PD, each FD+PD+ participant was asked about participant’s personal and family history of PD, specifically if other family members without FD, had been diagnosed or treated for PD or showed common symptoms of PD (resting tremor, shuffling gait, stooped posture, decreased arm swing, rigidity). PD clinical markers [MDS-UPDRS I-IV score off/on stage), subtype (tremor, akinetic-rigid, mixed, PIGD), Hoehn-Yahr stage, levodopa equivalent daily dose (LED)] were analysed.
Statistical analysis
Cohort characteristics and demographic data, for the two groups (FD+PD+ and their FD family members, FD+PD–), were reported using medians and inter-quartile range (IQR) for continuous variables and frequencies for categorical variables. As FD is X-linked, we stratified statistical analyses by gender. In order to perform a comparison between the two groups, due to the limited number of FD+PD+ subjects, we outlined their position in respect to the 95% confidence intervals estimated for the largest group (FD+PD–).
RESULTS
In 229 adult patients with FD due to the p.F113L mutation (157 patients aged ≥35 years; 101 patients aged ≥50 years; 61 patients aged >60 years), we found three patients with PD (prevalence of PD in FD: 3/229, 1.3%; 3/157, 1.9%; 3/101, 3%; 3/61, 4.9%, according to age cut-off).
FD+PD+ patients, one female (73 years old) (P1) and two males (60 and 65 years old) (P2 and P3), presented akinetic-rigid PD with a weak response to levodopa (6% – 36%) (LED: 300–500 mg) (Table 1) and no dyskinesias. The female patient (P1) presented PD 8 and 12 years later in comparison to male patients (P2 and P3, respectively), clearly offsetting as a more advanced stage of PD (Table 1), albeit without postural instability (HY = 2), presenting gait impairment and flexed posture refractive to levodopa (off/on MDS-UPDRS subcores: 3.10. (1/1); 3.13. (2/2)). Patient P1, besides p.F113L mutation, also carried a genetic variant of uncertain significance, in homozygous state, in the SLC30A10 gene (c.284C>T; p.Thr95Ile). This variant has been described in individuals manifesting with a syndrome of Hepatic Cirrhosis, Dystonia, Polycythemia and Hypermanganesemia; and individuals with juvenile-onset Dystonia and adult-onset Parkinsonism [22]. Patient P2 carried a genetic variant of uncertain significance, in heterozygous state, in LRRK2 gene (c.5687C>G; p.Ala1896Gly) that, to our knowledge, has not been reported yet. Nevertheless, in both P1 and P2 cases, no family history of parkinsonism was found in the previous generation. P3 patient had a negative test in the PD gene panel. 18F-DOPA PET scan revealed nigro-striatal dopaminergic presynaptic deficit in P1, P2 and P3 (Fig. 2). All FD+PD+ patients presented brain WML and the female patient (P1) had suffered a clinical stroke (Table 2, Fig. 3).
Demographics and clinical characteristics of FD+PD+ patients (P1, P2, P3) and their FD+PD–relatives
Data is presented individually for each of the three patients with Parkinson’s disease (PD) and Fabry disease (FD) and collectively for their relatives with FD in the three pedigrees (median/ interquartile range (IQR)) for continuous variables and number of patients or Yes/No for categorical variables). 1-two females and one male without MRI; 2-data available for 16 females; 3-data available for 9 females; 4- Data available for 11 females;.5-data available for 6 females; CRF, cerebrovascular risk factors; LED, Levodopa equivalent dose; ERT, enzyme replacement therapy; LVH, left ventricular hypertrophy; HBP, high blood pressure; NA, non-available data; MoCA, Montreal Cognitive Assessment; OT, other meds; PDQ-39, Parkinson’s disease Questionary-39; TIA, transient ischemic attack; UPRDRS, Unified Parkinson’s Disease Rating Scale in “Off” stage (without levodopa medication) and “On” stage (on levodopa medication); WML, white matter lesions. Reference values: enzymatic activity of α-galactosidase A on plasma 6–19 nmol/h/mL, and on leukocytes 36–80 nmol/h/mg; urinary Gb3 0.87 – 13μg/mmol creatinine; plasma Gb3 0.8–4.52 nmol/mL; plasma Lyso-Gb3 0–1.9 ng/mL.
Neurological and Neuroimaging findings in FD+PD+ patients and their FD+PD–relatives
Data is presented individually for each of the three patients (P1–P3) with Parkinson’s disease (PD) and Fabry disease (FD) and collectively (median/ interquartile range (IQR)) for continuous variables and number of patients Yes/No for categorical variables) for their relatives with FD in the three pedigrees stratified by gender; 1-two females without MRI; 2-one male without MRI; ACGS, Global cortical atrophy scale; CT, computerized tomography; MRI, magnetic resonance imaging; WML, white matter lesions.
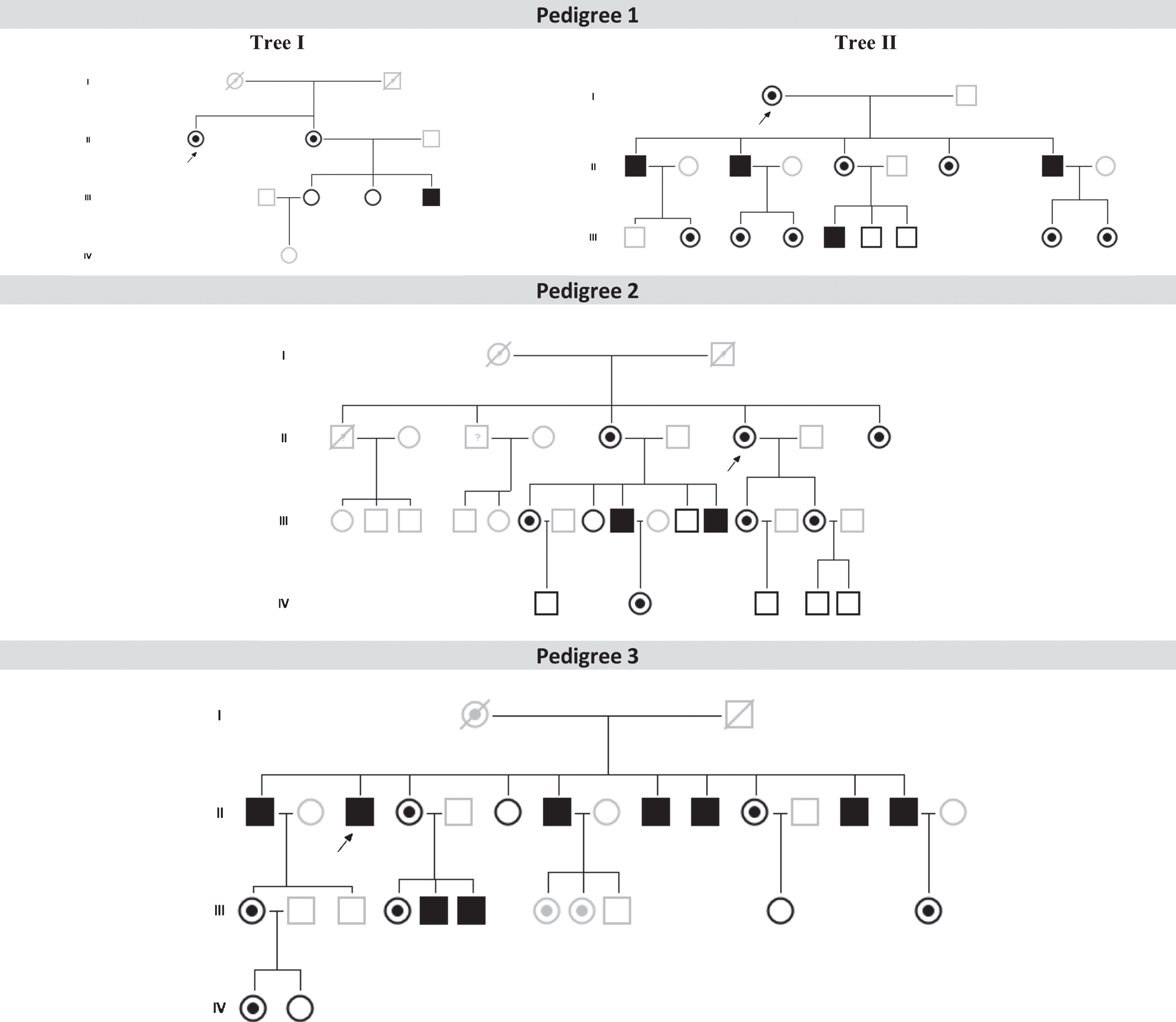
Family pedigrees from the three patients with Parkinson’s disease patients and Fabry disease (FD+PD+) (p. F113L mutation) and their relatives with or without FD. Index FD patients are marked with a black arrow (in Pedigree 1, individuals II-1 from Tree I and I-1 from Tree II are the same index patient). Patients with FD and PD: P1 (Pedigree 1, tree I, II-2); Patient 2 (Pedigree 2, III-9); Patient 3 (Pedigree 3, II-1). Dark symbols represent FD patients. White symbols represent relatives without FD. Grey symbols represent untested individuals.
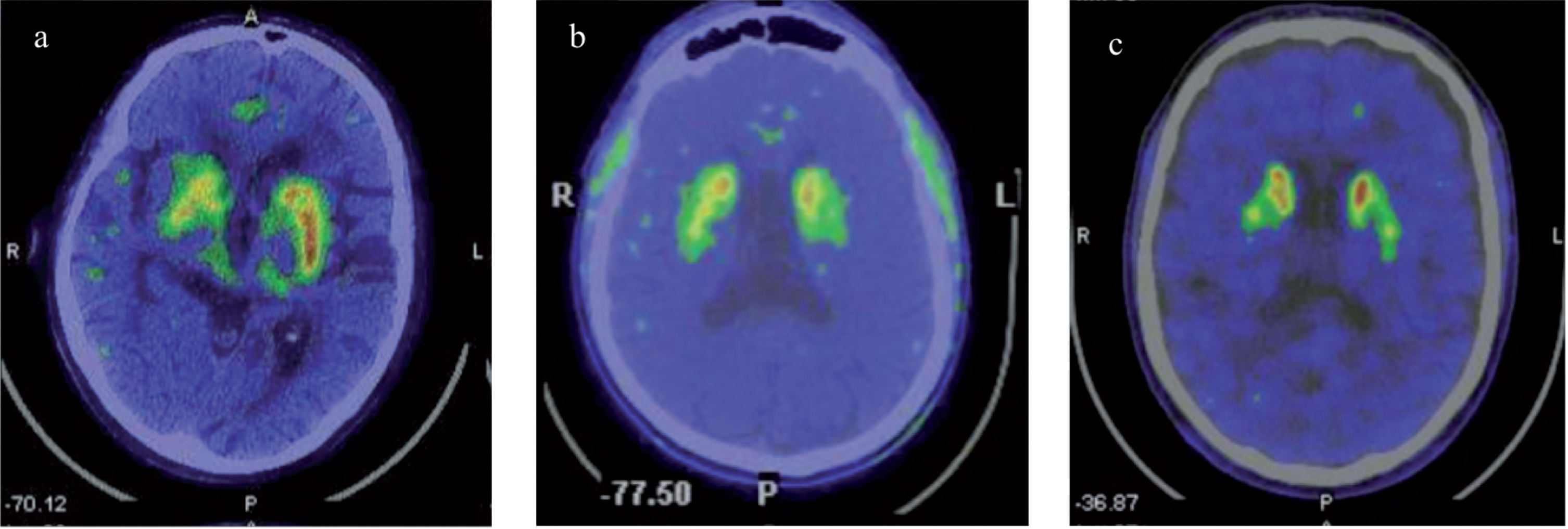
18F-DOPA PET scan performed in patients with Fabry Disease and Parkinson’s Disease revealed: a) reduced right striatal dopaminergic presynaptic deficit in patient P1; b) reduced left striatum dopaminergic presynaptic deficit in patient P2; c) and reduced right nigro-striatal dopaminergic presynaptic deficit in patient P3.
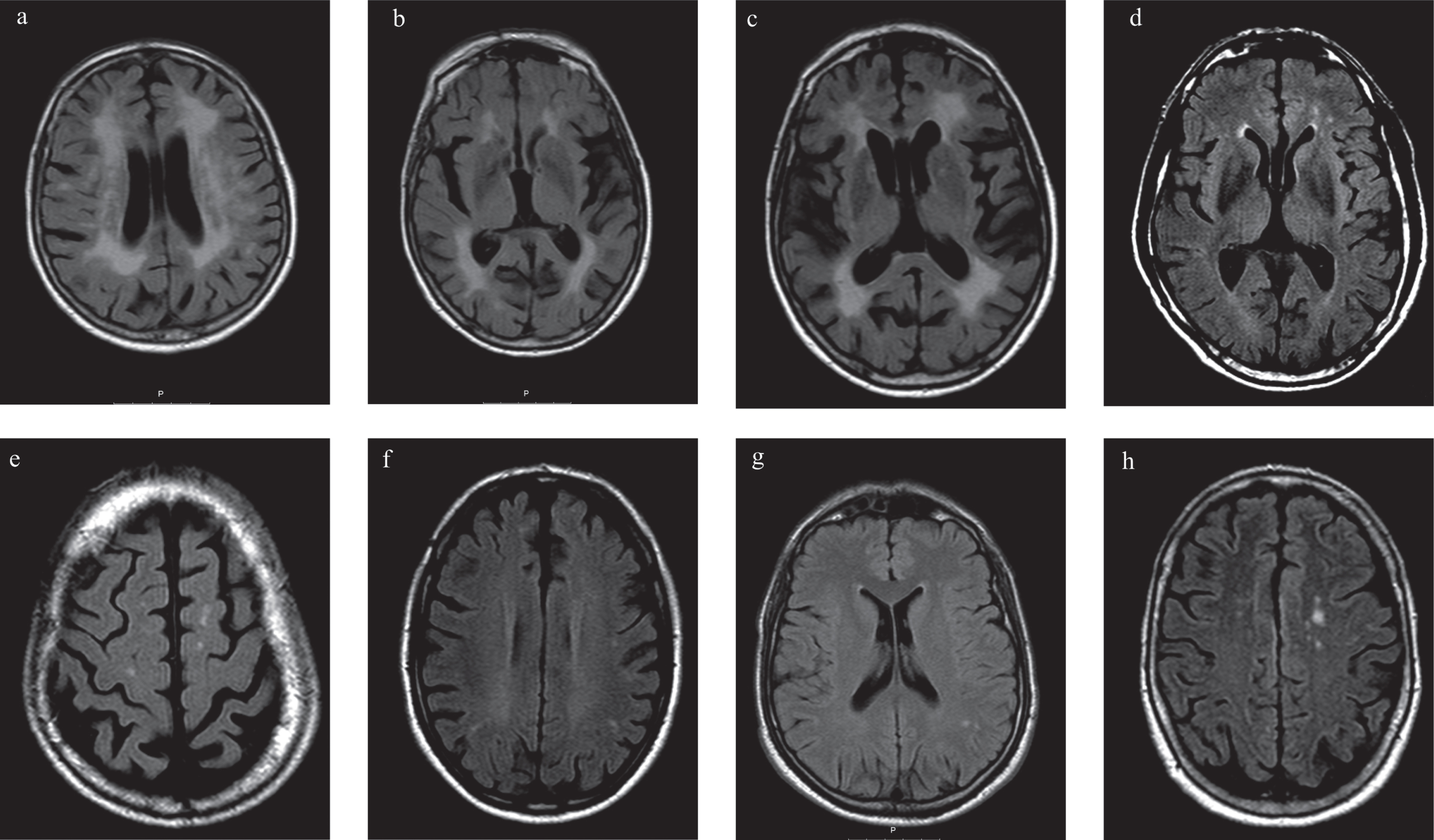
Brain magnetic resonance imaging (MRI) with axial Fluid Attenuated Inversion Recovery (FLAIR) of patients with Fabry disease (FD) and Parkinson’s disease (PD) (P1, P2, P3) and FD without PD (FD+PD–). a, b, c) MRI of P1; a) and b) Diffuse confluent bilateral hyperintensities in the periventricular and subcortical fronto-parietal white matter, suggesting leukoencephalopathy; c) Basal ganglia lacunar infarcts; d, e) MRI of P2, Frontal periventricular and subcortical white matter hyperintensities; f) MRI of P3, Frontoparietal periventricular and subcortical white matter hyperintensities; g) MRI of 43 years old, male with FD+PD–, White matter hyperintensities in the left parietal subcortical white matter; h) MRI of 75 years old, female FD+PD–patient, with multiple white matter hyperintensities in the subcortical frontoparietal regions and centrum semiovale.
FD+PD+ patients belonged to three different pedigrees, with each pedigree having 8 (P1), 9 (P2) and 13 (P3) family members with confirmed FD, totaling 30 FD patients (Fig. 1). From this cohort of 30 FD patients, 24 FD patients were older than 35 years old (5 (P1), 9 (P2) and 10 (P3)) and were included in our final analysis (16 females/8 males). Overall, the FD+PD+ patients had worse clinical severity of FD than their FD+PD- family members (Tables 1 and 2), clearly offsetting the upper IQR of the MSSI total score (all); and neurological (P1, P2); cardiovascular (all); and renal subscores (P1, P2) (Table 2).
Concerning α-GAL A enzymatic activity (Table 1, Fig. 4), P1 female patient presented plasma α-GAL A activity (P1:8.0 versus Females FD+PD–: 7.95 (median) (nmol/h/mL)) and leukocytes α-GAL A activity (P1:51.0 versus Females FD+PD–: 30.5 (median) (nmol/h/mg)) in the same normal range of its female counterparts. In contrast, male FD patients had significant lower and almost absent α-GAL A activity, with P2 and P3 male patients portraying plasma α-GAL A activity in the same range as their male counterparts (P2, P3:0–0.7 versus Male FD+PD–: 0 (median) (nmol/h/mL)). Still, P2 and P3 had slightly higher α-GAL A activity in leukocytes (P2, P3:2.2-3 versus Male FD+PD–: 1.0 (median)(nmol/h/mg)).
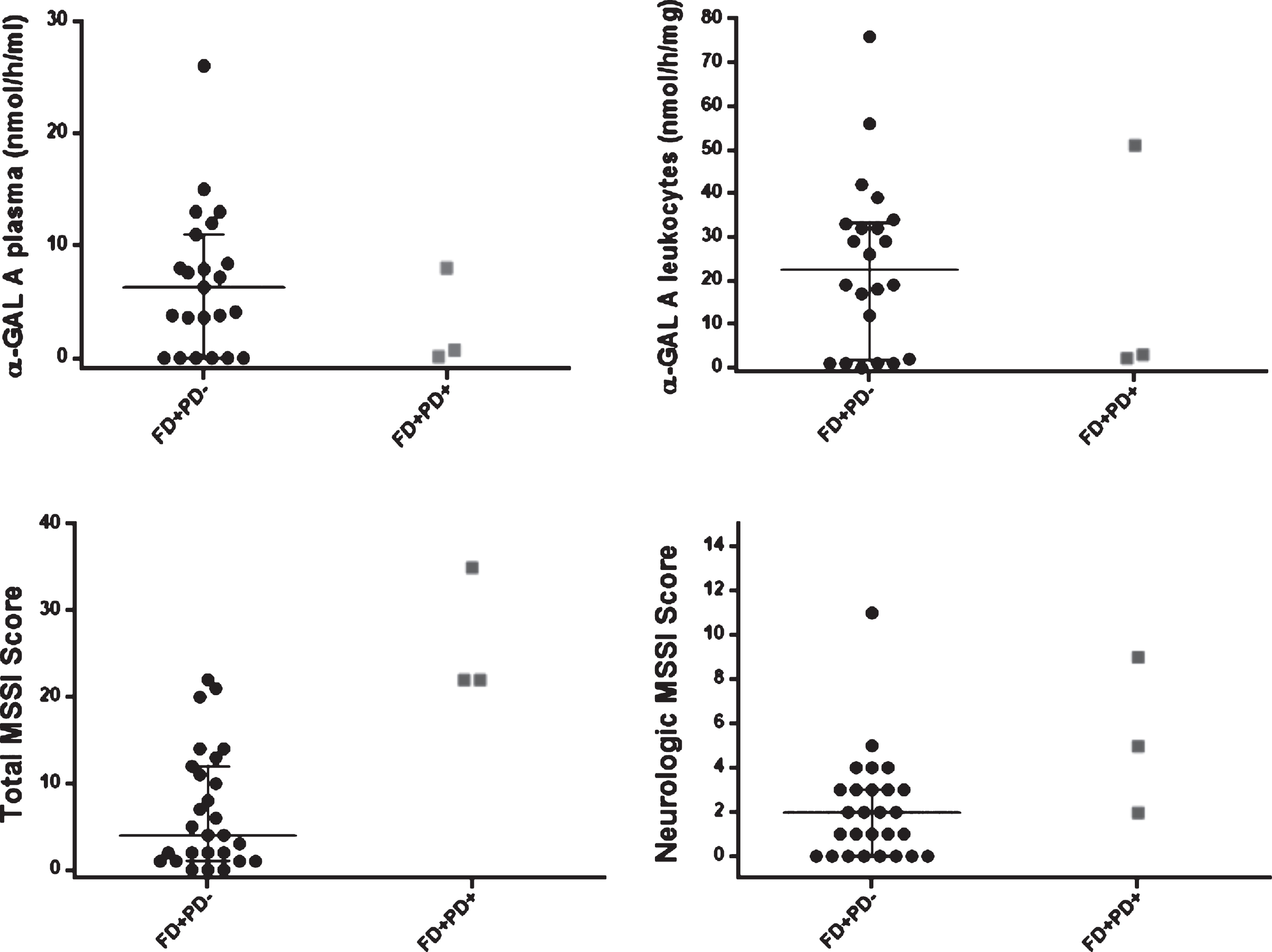
Scatter dot plot, with median and IQ intervals (25–75%) for α-GAL A enzymatic activity (plasma, leukocytes), Mainz Severity Scale Index (MMSI) (Total and Neurological Score) in patients with FD (FD+PD–) (black circle dots) and FD and PD (FD+PD+) (grey square dots). In the FD+PD+ group, the upper dot is P1 female patient. Reference values: enzymatic activity of α-galactosidase A on plasma 6–19 nmol/h/mL, and on leukocytes 36 –80 nmol/h/mg; urinary Gb3 0.87 –13μg/mmol creatinine; plasma Gb3 0.8 –4.52 nmol/mL; plasma Lyso-Gb3 0 –1.9 ng/mL.
Concerning FD biomarkers, P3 male patient had lower value of Plasma Lyso-Gb3 (P3:5.76 versus. Males FD+PD–: 9.2 (median)(ng/mL)), which is most likely explained by the fact of being under enzyme replacement therapy (ERT), agalsidase beta (70 mg iv, every other week) for 24 months, since the age of 63 years old, 1 year previous to PD diagnosis.
Notwithstanding other cardiovascular risk factors (e.g., diabetes, hypertension) that were also frequent (Table 2), brain WML (100% of FD+PD+ patients, 38% of FD+PD–females and 60% of FD+PD–males) and small vessels ischaemic lesions (100% of FD+PD+ patients, 15% of FD+PD- females and 20% of FD+PD- males) were widely present in FD patients, with higher prevalence in FD+PD+ patients.
DISCUSSION
To the best of our knowledge, this is the first study surveying PD in a large cohort of FD patients with the p.F113L mutation. We found a higher prevalence of PD in FD patients (1.3% in the total cohort of adult FD patients and 3.0% in the subgroup of FD patients aged ≥50 years), when compared to the 0.24% adjusted prevalence of PD in community-dwelling Portuguese population, aged ≥50 years [23]. With respect to previous reports, we have found at least two common similarities, which can be highlighted as characteristic of FD with PD: higher burden of cardiac and cerebrovascular lesioning.
Orimo et al. [12] reported a 68-year-old male with FD (Gb3 deposits in cardiac and kidney autopsy, but without genetic investigation) presenting an atypical form of parkinsonism, beginning at the age of 63 years old, with axial signs (gait and postural instability), mild symmetrical rigidity, and pyramidal signs (mild right hemiparesis, generalized hyperreflexia and positive Babinski sign on both sides). Brain MRI showed multiple T2 hyperintensities in the basal ganglia and deep white matter regions, suggesting the possibility of vascular and/or neuronal dysfunction in FD [24]. Buechner et al. [13] reported a 57-year old woman with FD due to the p.R301P GLA gene mutation, with low leukocyte α-Gal A activity (8 nmol/mg per hour), cardiac (left ventricular hypertrophy) and kidney impairment (microalbuminuria of 31.4μg/mL), presenting mild parkinsonism beginning at the age of 46 years old. Even though presenting a good response to levodopa, the patient developed motor complications four years later, complicated by axial signs (gait impairment with freezing). Brain MRI showed extensive leukoencephalopathy with multifocal ischemic lesions, including in the head of the right caudate nucleus. These two distinctive clinical reports share these similar findings: cardiac and renal impairment; high burden of cerebrovascular lesioning and parkinsonism with precocious axial impairment.
In our cohort, even though the two FD+/PD+ male patients (P2-P3) suffered a mild parkinsonism and LED dose can be further increased, the female patient (P1) presented, in less than 18 months, a moderate-severe parkinsonism with very low response to levodopa. This refractoriness to levodopa can be explained by concomitant cerebrovascular lesions affecting directly nigro-striatal neural structures (P1 patient with basal ganglia and brainstem infarcts), and/or indirectly subcortical fibers (P2-P3 patients).
As we previously reported, the p.F113L GLA gene mutation is associated with a high burden of cardiac and CNS manifestations [25, 26]. Hearing loss was a common manifestation and age-matched hearing thresholds by frequency are worse in FD than in the general population [18, 26]. One possible explanation is that the vascular peripheral lesioning could correlate with higher CNS microvascular burden (hearing loss was correlated with microalbuminuria). CNS complications in FD have been mostly attributed to cerebral vasculopathy, Gb3 storages within neurons [27], cerebrovascular risk factors [28], and genetic factors [29], increasing the likelihood of cerebrovascular disease. Even though statistical inference from three patients is considerably limited, it is striking that all FD+PD+ patients had small vessel ischaemic brain lesions. This observation raises the question whether cerebrovascular lesions, highly frequent in FD [30], increase by themselves the risk of PD and/or instead alter its phenotype. Also, even though the criteria for pure Vascular Parkinsonism are still controversial [31, 32], cerebrovascular lesions probably alter the natural history of PD, conditioning a more severe, axial phenotype of PD [33], as we may have observed in our study.
It is still open to discussion how lower α-GAL A activity (plasma and leukocytes) correlates with α-synuclein abnormal degradation in sporadic PD or in PD patients carriers of a lysosomal mutation in GBA gene. Also, to our knowledge, there is no previous literature concerning biochemical analysis in PD patients carriers of a GLA gene mutation and concomitant FD. In Alcalay et al. work, when including sporadic PD (n = 468) and GBA (n = 110) and LRRK2 G2019S carriers (n = 42) with PD, but without FD, PD patients presented significantly lower α-GAL A enzymatic activity in dried blood spots (PD: 2.89±1.41 versus Controls: 3.10±1.68μmol/L/h; p = 0.040) [10]. This observation remained even stronger when considering only the non-carriers sporadic PD group (2.85 versus 3.12μmol/L/h, p = 0.018). Yet, when Alcalay et al. stratified analysis by gender, contrarily to what was found in females, sporadic male PD patients did not present lower α-GAL A activity in comparison to controls (2.90 versus 3.17μmol/L/h, p = 0.123) [10]. Also, in PD male patients with GBA gene mutations (n = 10) [10], α-GAL A activity was not significantly different from the one in sporadic PD. In contrast, in male PD patients with LRRK2 G2019S mutation, the α-GAL A activity was higher than in non-carriers (mean normalized α-GAL A activity: 1.15 vs. 1.03, p = 0.032). Interestingly, in our study, in comparison to their male counterparts of FD+PD group, our FD+PD+ male patients portrayed higher leucocyte α-GAL A activity (Table 2, Fig. 4). One possible biological explanation for discrepancy in different studies, already presented by Alcalay et al., is that α-GAL A activity may vary (increase or decrease) as a compensatory mechanism in response to other upstream cellular dysfunctions in the endolysossomal system.
Study strengths and limitations
Our study approach has substantial strengths in comparison to previous literature, as it accounts for a prospective screening for PD in patients with an already established diagnosis of FD, complemented with biochemical and neuroimaging analysis.
Our description of FD patients with PD and respective pedigrees has several limitations. Screening for the major known PD genes in these three patients led to the identification of a variant in the SLC30A10 gene (P1) and a variant in the LRRK2 gene (P2), both of uncertain significance, and further studies are needed in order to establish the clinical significance of these variants. Even though all 229 patients were prospectively evaluated, the search for PD diagnosis was not systematic, since clinical diagnosis was made in general neurological approach. On the other hand, some FD patients are still not old enough to manifest clinical motor signs of PD, further hampering any inference for the risk of developing PD in carriers of the p.F113L GLA gene mutation. Still, we found a prevalence of 3% in patients aged ≥50 years, which is considerably above the 0.24% adjusted prevalence of PD in community-dwelling Portuguese population, aged ≥50 years [23].
Future directions
The prodromal phase of PD begins decades before its clinical onset (when more than 60% of nigrostriatal loss is already present), namely some non-motor symptoms (TREND and PRIPS studies) [34]. Lohle et al. [35] found that, while FD patients (60 females and 50 males with GLA gene mutations) commonly present impaired motor function and various non-motor symptoms (bradykinetic motor phenotype, including slower gait and lower hand speed), classic prodromal features of PD (hyposmia/anosmia, autonomic dysfunction, REM Sleep Behaviour Disorder) were absent. As such, it would be most relevant to perform 18-F DOPA PET in non-PD FD patients as well as to perform a prospective follow-up, from a young age, with additional PD clinical and neuroimaging markers (e.g., neuromelanin MRI). Also, in order to ascertain the impact of vascular lesions in PD phenotype, a post-synaptic dopamine PET imaging (e.g., raclopride-PET) would confirm or exclude striatal neuronal degeneration. As FD+PD+ patients carry the same p.F113L mutation and share a common ancestry, the possibility of co-segregation of other genetic alterations and different diseases deserves proper molecular extensive genetic analysis.
Conclusion
In summary, this study showed a higher prevalence of PD in FD patients than in the general population (1.3% in the total cohort of adult FD patients and 3.0% in the subgroup of FD patients aged ≥50 years), further highlighting the importance of the autophagy-lysosomal pathway in PD. All FD+PD+ patients presented an akinetic-rigid PD with weak response to levodopa as well as worse clinical severity of FD, brain WML and high cerebrovascular burden. Further studies will be needed to understand how much of this PD phenotype is due to deposition of Gb3 versus cerebrovascular lesions in the nigro-striatal network. Although the discussion of Vascular Parkinsonism and overlapping syndromes of Vascular Parkinsonism with PD is controversial, it is plausible, at this moment, to foresee that cerebrovascular burden will be a very important co-variable determining the final phenotype of PD in FD patients. Moreover, although ERT has not been shown to cross the blood-brain barrier [36], prospective follow-up will demonstrate if its cardiovascular and renal benefits have meaningful impact in PD history. Finally, future studies will be needed to corroborate if, upon PD neurodegeneration, patients activate a compensatory mechanism of increased α-GAL A activity as it was observed in LRRK2 carriers.
CONFLICT OF INTEREST
The authors have no conflict of interest to report
Footnotes
ACKNOWLEDGMENTS
The authors are grateful for the clinical support provided by all colleagues working in the Reference Centre on Lysosomal Storage Disorders, and in the Neurology and Cardiology Departments, Hospital da Senhora da Oliveira, EPE, Guimarães.