
Abstract
Background:
Opicapone is a catechol O-methyltransferase (COMT) inhibitor indicated for use as adjunct to levodopa therapy in patients with Parkinson’s disease (PD) and motor fluctuations.
Objective:
To characterize the safety and tolerability of adjunct opicapone (25 and 50 mg) in a pooled population of levodopa-treated PD patients who participated in the opicapone Phase-3 clinical program.
Methods:
Patient-level data (placebo, opicapone 25 mg and 50 mg) from the BIPARK-1 and BIPARK-2 double-blind and open-label studies were combined.
Results:
Pooled analyses included 766 patients from the double-blind studies and 848 patients from the open-label studies. In the double-blind studies, 63.3% of opicapone-treated patients reported treatment-emergent adverse events (TEAEs) versus 57.2% in the placebo group. The most common TEAEs reported in the opicapone group compared to placebo were dyskinesia, constipation and insomnia. The incidence of serious TEAEs was similar across opicapone and placebo groups (3.5% versus 4.3%, respectively). Overall, 71.3% patients treated with open-label opicapone reported at least one TEAE; most occurred within the first 2 months of the open-label studies, and then decreased thereafter. Throughout the Phase-3 clinical program, there were no serious AEs suggestive of hepatic toxicity, and the incidence of gastrointestinal disorders such as nausea and diarrhea remained low (<2%). There were no relevant changes in laboratory parameters including liver enzymes, vital signs, physical or neurological examinations, or ECG readings.
Conclusions:
Long-term use of opicapone once-daily over 1-year at doses of 25 mg or 50 mg was generally safe and well tolerated, supporting its clinical usefulness in the management of PD motor fluctuations.
INTRODUCTION
Opicapone is a third generation, catechol O-methyltransferase (COMT) inhibitor that was approved in 2016 by the European Union for use as adjunctive therapy to preparations of L-DOPA/DOPA decarboxylase inhibitors (DDCIs) in adult patients with Parkinson’s disease (PD) and end-of-dose motor fluctuations who cannot be stabilized on those combinations [1]. The preclinical development of opicapone was based on a rational, analogue-based research approach and was specifically designed to reduce the risk of toxicity and improve peripheral tissue selectivity compared with other COMT inhibitors [2].
The efficacy, safety and tolerability of opicapone given once daily as adjunct to levodopa has been established in two large randomized clinical trials (BIPARK-1 [3] and BIPARK-2 [4]) and their associated open-label extension studies [4, 5]. Taken together, they provide safety data for up to 1 year for 848 patients. Similarities in the design of the two double-blind, randomized, placebo-controlled studies and their extensions permit a pooled analysis to provide further information on the safety and tolerability of opicapone and were an important component of the approval process in Europe [6]. Moreover, Olanow et al recently proposed that adverse event (AE) safety reporting in PD trials should also include information regarding the temporal profile of the AE’s reported, particularly dyskinesia [7]. The aim of this pooled analysis was to provide a comprehensive overview of the safety of opicapone, including more information on the temporal profile of potentially treatment-related AEs, in a pooled population of levodopa-treated patients experiencing motor fluctuations.
METHODS
BIPARK-1 [3] and BIPARK-2 [4] were randomized, double-blind placebo-controlled studies, full details of which have been published. Both were followed by open-label extension studies which followed patients for up to a year of treatment [4, 5]. All four studies were conducted in accordance with Good Clinical Practice and the provisions of the International Conference on Harmonization and were sponsored by BIAL - Portela & Ca, S.A.
Study designs
In brief, both studies recruited adult (aged 30–83 years) patients with a 3-year diagnosis of idiopathic PD and a Hoehn and Yahr stage between 1–3 during ON. For the double-blind studies, eligible patients had to be receiving 3–8 daily doses of levodopa for at least 1 year and experiencing end-of-dose motor fluctuations with ≥1.5 hours of OFF-time per day, excluding pre-dose morning akinesia. Patients with clinically significant and unstable cardiovascular disease or psychiatric illnesses (including major depression, dementia, impulse control disorders, and suicide ideation) or with relevant liver disease or transaminase levels greater than 2x the upper normal limit (ULN) were excluded. Concomitant stable treatment for PD was allowed, with the exception of tolcapone, apomorphine (withdrawn ≥1 month before screening), and entacapone (other than that supplied for BIPARK-1).
Both double-blind studies assessed the safety and tolerability of opicapone 25 mg and 50 mg versus placebo [3, 4]. However, BIPARK-1 was an active controlled study and included an additional entacapone arm, and a low dose (5 mg) opicapone arm (not relevant in the present analyses) [3]. The open-label studies began the day after completing the double-blind phase, and continued until the patient had completed 52 weeks of open-label treatment. Patients started open-label treatment with the 25 mg dose, and could be up-titrated to 50 mg if greater symptomatic control was required. In cases dopaminergic adverse events (AEs), the levodopa dose was to be adjusted first, followed by opicapone down-titration in those with persisting AEs [4, 5]. Adverse event reporting was collected at every visit throughout the studies. Laboratory and vital sign tests were performed at screening, baseline, day 14 or 21 and monthly thereafter up to end-of-study/early discontinuation visit.
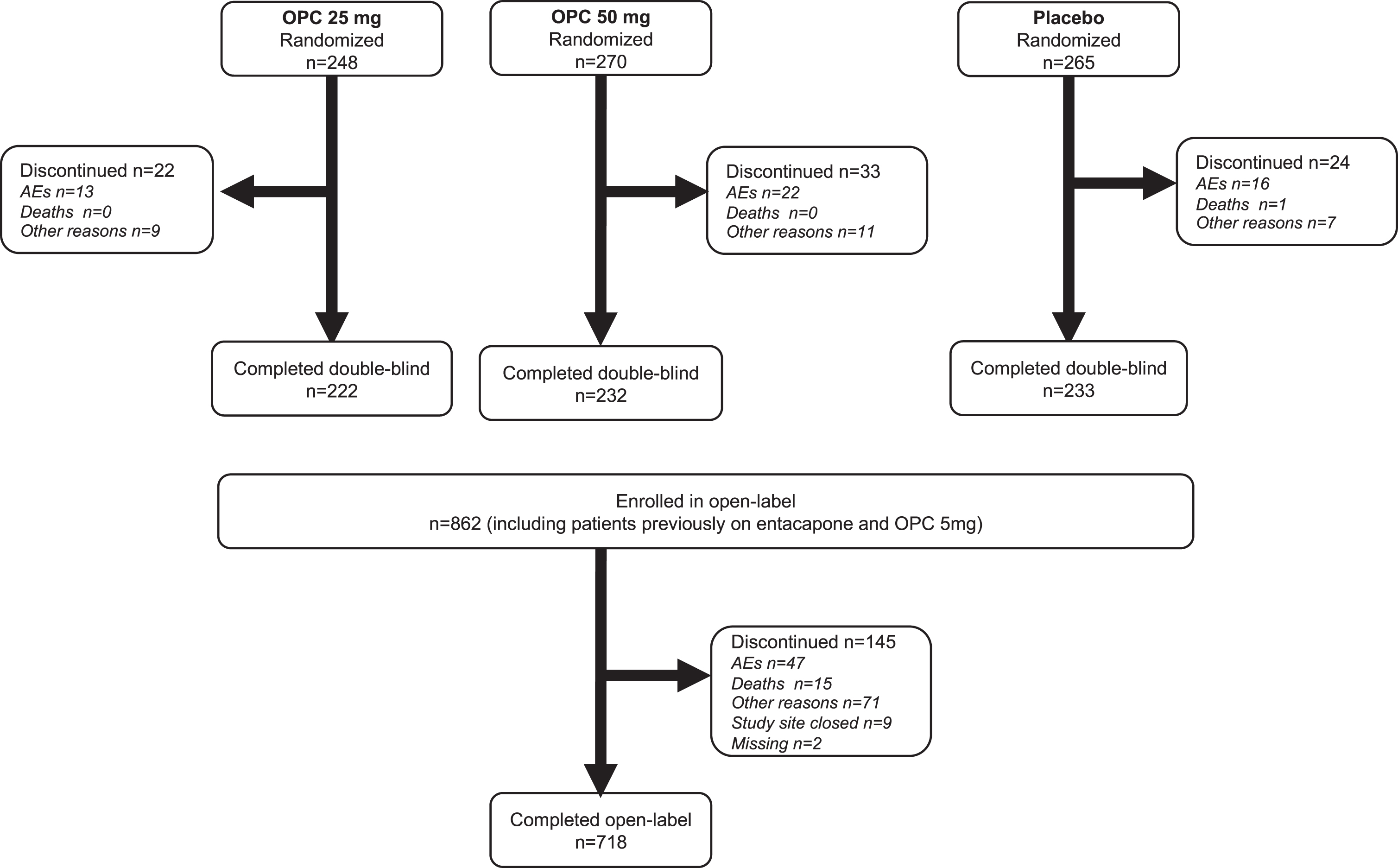
Patient disposition.
Analysis
Patient-level data from matching treatment arms in the double-blind studies (placebo, opicapone 25 mg and opicapone 50 mg) and all treatments from their open-label extensions was combined and analyzed using descriptive data. Safety was assessed by incidence of treatment-emergent adverse events (TEAEs), laboratory parameters, ECGs and vital signs.
In accordance with European Medical Agency guidelines [8], we predefined the following TEAEs as being of special interest to COMT inhibitors or Parkinson’s disease: psychosis, depressive events, sleep disorders, orthostatic hypotension, serious hepatic events, severe diarrhea, myocardial infarction, increased creatine phosphokinase (CPK), impulse control disorders and/or melanoma. For the double-blind studies, we also performed an analysis of changes in hepatic laboratory parameters, including aspartate and alanine transaminases (AST and ALT), alkaline phosphatase (ALP), total bilirubin (TBL) and gamma-glutamyl transferase (GGT) using the last observation carried forward (LOCF) approach to account for missing data.
Finally, we performed post-hoc subgroup analysis comparing the double-blind safety of opicapone in the elderly (aged ≥70 years) and non-elderly (<70 years old) populations.
RESULTS
Patient disposition and baseline characteristics
Of the 1027 patients randomized to the two double-blind studies, 766 patients took a dose of relevant study medication (509 patients received opicapone 25 mg or 50 mg, 257 received placebo) and were included in the double-blind analysis (Fig. 1). The open label analysis included all 848 patients enrolled to open-label treatment who took at least one dose of study medication.
Baseline characteristics at the start of the double-blind phase are shown in Table 1. Overall, the majority (83%) of patients received polypharmacy (levodopa plus at least one other PD medication) for their parkinsonian symptoms. Most patients (>88%) had a co-morbid illness, the most common were musculoskeletal and connective tissue disorders (e.g. osteoporosis and osteoarthritis) and psychiatric disorders (e.g. anxiety, depression).
Baseline characteristics
Double-blind phase
In the double-blind studies, 63.3% (n = 322) of patients in the opicapone groups reported a TEAE compared to 57.2% (n = 147) in the placebo group. Most TEAEs were mild or moderate in intensity. The only TEAEs which occurred in ≥5% of patients in either opicapone group and were more common with active treatment versus placebo were: dyskinesia, constipation, insomnia and dry mouth (Table 2). Most TEAEs occurred early (Fig. 2) and were mostly transient. Dyskinesia was the most frequent TEAE, but the incidence of severe cases was low (opicapone 1.2% vs. placebo 0.8%) and most dyskinetic events occurred within the first 4 weeks of opicapone treatment (13.9% of opicapone patients (n = 71) had dyskinesia as a TEAE during Weeks 1–4). Figure 2 includes the temporal profile of dyskinesia events during the placebo-controlled phase. At Week 1, 9.07% of patients in the 50 mg group reported dyskinesia and this reduced to just 0.44% at Week 14 (a relative reduction of 95.15%). By contrast, there was no clear pattern for onset of constipation, although rates per week were consistently low.
TEAEs in the double-blind studies
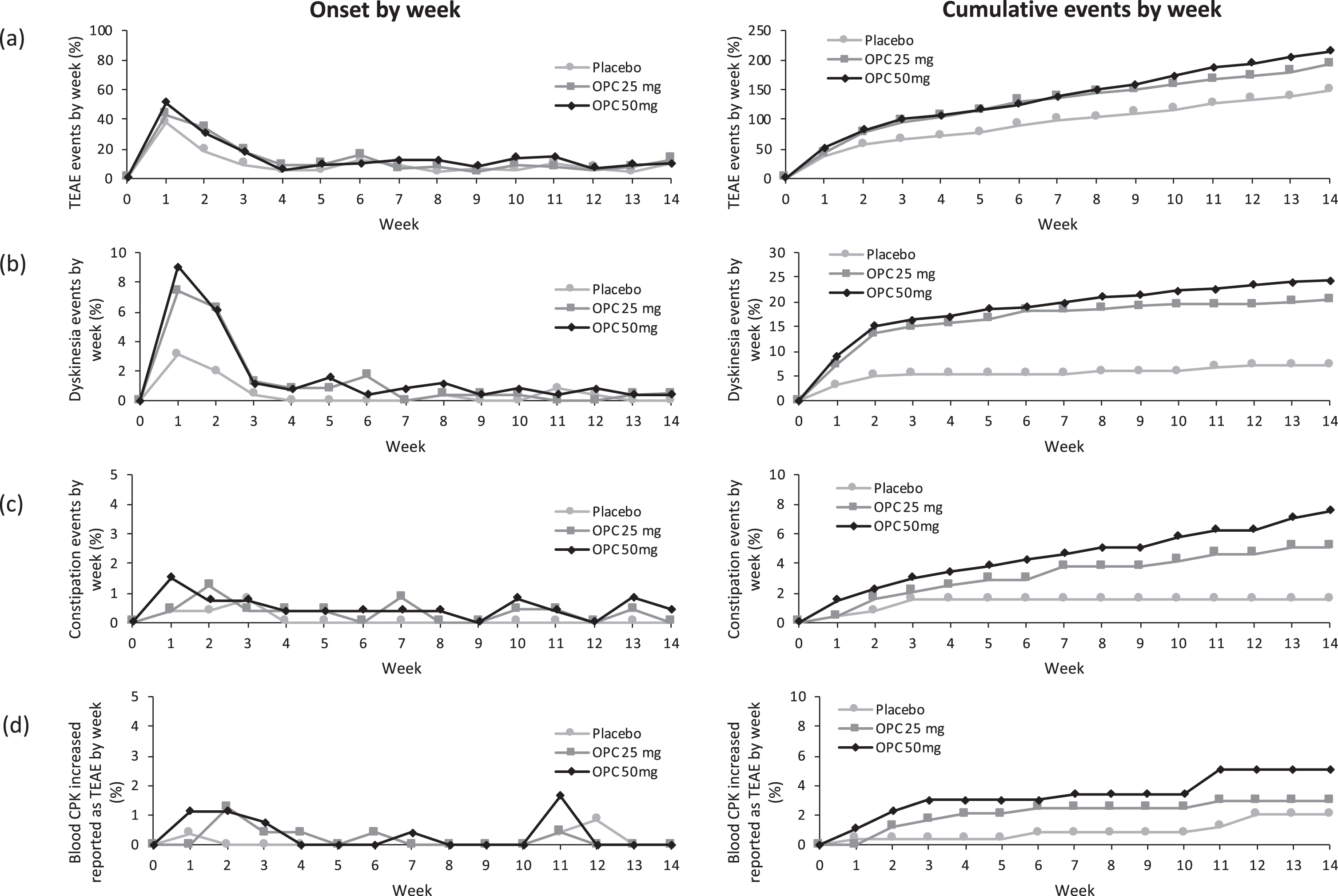
Temporal profile (onset and cumulative rates) of adverse events during the placebo-controlled phase (a) overall treatment emergent adverse events (b) dyskinesia (c) constipation and (d) increased creatine phosphokinase. Note. Graphs show number of events and any given subject may have reported >1 event. OPC = opicapone.
TEAEs that could possibly be dose-related (i.e. those for which the incidence increased with increasing opicapone dose) were mostly as expected with dopaminergic therapy (e.g. dyskinesia, constipation, hallucinations). Increased CPK levels were also potentially dose-related and were mostly reported in the first 3 weeks (Fig. 2). Thereafter, with the exception of Week 11, there were no reports of increased CPK as a TEAE at any other timepoint. Most cases of increased CPK levels were of mild intensity, transient and resolved without action.
In addition, pooled analysis revealed infrequent, but potentially dose-related, cases (50 mg vs. 25 mg vs. placebo) of: leukopenia (1.5% vs. 0% vs. 0.4%), urinary tract infection (3.8% vs. 1.6% vs. 0.8%), crystal present in urine (1.5% vs. 0.4% vs. 0%), and memory impairment (1.1% vs. 0.4% vs. 0%) that were not expected with opicapone therapy. None of the events of urinary tract infection or crystal present in urine were considered to be related to study medication. Leukopenia was considered potentially treatment-related in two patients treated with opicapone 50 mg and one patient treated with placebo (i.e. treatment-related AE rate of 0.4% in both groups). For memory impairment, only one patient had an event considered related to opicapone (50 mg).
The incidence of serious TEAEs was similar across opicapone and placebo groups (3.5% in combined opicapone groups vs. 4.3% with placebo). Thirteen patients (4.9%) in the 50 mg opicapone group had at least one serious AE. Serious events included: constipation, inguinal hernia, duodenitis, nausea, acute cholecystitis, coronary heart disease, acute pyelonephritis, fall, head injury, prostate biopsy, basal cell carcinoma, dyskinesia, febrile delirium, cystocele, pleural effusion, pulmonary embolism (all n = 1). Five patients (2.0%) in the 25 mg opicapone group had a serious AE including: osteoarthritis, Bowen’s disease, dyskinesia, acute renal failure, and urinary retention (all n = 1). Dyskinesia was the only serious AE considered to be treatment-related (n = 1 in both the opicapone 25 mg and 50 mg groups).
Overall ten patients (2.0%) discontinued due to dyskinesia, followed by vomiting (n = 4, 0.8%), dizziness (n = 3, 0.6%), visual hallucinations (n = 3, 0.6%) and nausea (n = 3, 0.6%). There were no reports of serious hepatic events (no cases of Hy’s law), severe diarrhea, myocardial infarction or melanoma with opicapone treatment. However, two serious hepatic events were reported in the placebo group (acute hepatitis and hepatic enzyme increased). Other AEs of special interest were infrequent (total opicapone vs. placebo): psychosis (5.5% vs. 1.2%), sleep disorders (8.4% vs. 5.1%), orthostatic hypotension (1.2% vs. 0), impulse control disorders (0.4% vs. 0), depressive events (4.1% vs. 2.3%), CPK increase (3.9% vs 1.9%). There were no clinically relevant differences in vital signs, ECG readings or laboratory parameters, including hepatic laboratory parameters between groups. In particular, the incidence of potential clinically important hepatic laboratory values was low and similar between opicapone and placebo groups (Supplementary Figure e1).
Subgroup analysis demonstrated a comparable incidence of TEAEs in elderly patients ≥70 years treated with opicapone to age-matched placebo patients (66.0% vs. 63.8%). The TEAEs occurring more frequently in elderly patients compared to patients <70 years (and more common with opicapone than placebo) were: hallucinations (4.6% vs. 0.1%), visual hallucinations (3.8% vs. 0.1%) and decreased weight (4.6% vs. 1.1%). The incidence of serious TEAEs in patients ≥70 years was lower in opicapone treated patients compared to age-matched placebo patients (7.2% vs. 10.1%).
Open-label phase
As per protocol, all patients began open-label treatment with opicapone 25 mg. Overall, 439 (51.8%) patients completed the study on the 50 mg opicapone dose. Only 31 (3.7%) of patients reduced their opicapone dose to 5 mg.
Overall, 605 (71.3%) patients treated with open-label opicapone reported at least one TEAE. Table 3 shows TEAEs in at least 2% of patients in the open-label extensions. As in the double-blind phase, most TEAEs were of mild or moderate intensity and 5.5% of patients (n = 47) prematurely discontinued due to AEs. The majority of TEAEs occurred within the first 2 months of the open-label studies (256 events occurred during Weeks 1–4 and 135 events during Weeks 5–8), and then decreased thereafter (30–98 events per month).
TEAEs in at least 2% of patients in the open-label extensions; n (%)
A total of 87 (10.3%) patients experienced at least one serious AE. Of these, potentially related SAEs were reported in 14 (1.7%) patients and included malignant melanoma (n = 2) and (all n = 1) abnormal behavior, aggression, agitated depression, atrioventricular block complete, cerebral hemorrhage, confusional state, craniocerebral injury, dementia, depression, diarrhea, dyskinesia, epilepsy, auditory hallucination, head injury, jealous delusion, orthostatic hypotension, fractured skull and wrist fracture. Fifteen deaths occurred, 14 of which were considered unrelated to study medication (death [unknown cause], sudden death, cardiovascular insufficiency, myocardial infarction, multi-organ failure, pneumonia, septic shock, craniocerebral injury, skull fractured base, metastases to spine, small cell lung cancer, lung disorder, pulmonary embolism and embolism). One death (cerebral hemorrhage after traumatic brain injury) was considered possibly related to study medication.
There were no serious AEs suggestive of hepatic toxicity, and the incidence of gastrointestinal disorders such as nausea and diarrhea remained low (<2%). There were no relevant changes in laboratory parameters including liver enzymes, vital signs, physical or neurological examinations, or ECG readings. There was no indication of an increase in suicidality or in the incidence of impulsive disorders over the year of open-label treatment with opicapone.
DISCUSSION
This pooled analysis combines data from two similarly designed pivotal studies with their open-label extensions and provides a comprehensive overview of the safety and tolerability of opicapone as adjunct to levodopa in fluctuating PD for up to one year of treatment. Most adverse events were of mild or moderate severity, and in general were transient and manageable, as indicated by the low discontinuation rate.
The rationale for using COMT inhibitors is to increase levodopa bioavailability, and so it is predictable that the most common AEs with opicapone therapy are dopaminergic in nature. Across both double-blind studies, the most common TEAEs reported in the opicapone group compared to placebo were the dopaminergic events of dyskinesia, constipation, insomnia, dry mouth and dizziness, as well as increased CPK. Dopaminergic AEs tend to occur shortly after introducing a COMT inhibitor and are usually transient, their reduced frequency being coincident with the decrease in total daily levodopa dose. During open-label treatment, the incidence of most of these dopaminergic TEAEs remained low and similar to that seen in the double-blind pivotal studies.
Dyskinesia was consistently the most frequently reported TEAE considered related to the study drug. Temporal analysis shows that rates of dyskinesia were highest in the first few weeks of opicapone treatment, and generally decreased over time as patients had their levodopa doses adjusted. Previous in depth analyses of the BIPARK-1 study have shown that (i) most dyskinesia events occurred in patients who were already experiencing dyskinesia at baseline, and (ii) although dyskinesia rates were higher for opicapone 50 mg versus entacapone 200 mg during the first 2 weeks of treatment, rates were similar following levodopa dose adjustment [3, 9]. Due to methodologic necessity, the pivotal studies did not allow levodopa dose reductions during the last 12 weeks of the study, but this would not be an issue in clinical practice where levodopa dose reductions can be readily made according to the clinical condition of the patient [1]. In this respect, the simple once-daily dosing of opicapone enables an easier titration of the levodopa dose compared to the complexity of reducing doses of the combined levodopa/carbidopa/entacapone pill.
Levodopa is considered the drug of choice for elderly patients [10], and it is therefore of practical importance that opicapone appears to be equally well-tolerated in elderly (aged 70+ years) and younger patients (<70 year old). Opicapone was well tolerated in the face of polypharmacy and co-morbidity; most patients in the opicapone Phase 3 program (88–97%) had a comorbid illness, and the majority were already receiving a variety of other adjunct antiparkinsonian medications (in addition to their baseline levodopa).
Adverse events of special interest for COMT inhibitors include hepatic and gastrointestinal safety. Hepatic safety has been the most important limitation of tolcapone in routine practice [11] and has prevented other COMT inhibitors coming to market. In these pooled analyses, there was no apparent increase in hepatic disorders with opicapone and opicapone (25 mg or 50 mg once-daily) did not cause any relevant change in hepatobiliary laboratory parameters. The incidence of potential clinically important liver enzyme values was low (<1% for any parameter) and comparable to placebo. Similarly, it has been suggested that gastrointestinal problems such as severe diarrhea may be a class effect of COMT inhibition [12]. Whereas severe diarrhea was the most frequent cause of discontinuation in the tolcapone (up to 10% [12]) and entacapone (3-4% [13, 14]) trials, no patient in the opicapone groups withdrew due to diarrhea in the double-blind studies, and only one patient reported a serious adverse event of diarrhea in the open-label extensions (this case resolved without a dose change). These data argue against diarrhea as a class effect. It is also noteworthy that opicapone is not associated with potentially embarrassing urine discoloration or orange staining of teeth, hair or nails associated with entacapone administration [12]. Indeed, placebo pills in the BIPARK-1 study had to maintain blinding by including riboflavin to mimic the urinary discoloration caused by entacapone [3].
The main strength of these analyses are the large numbers of patients included, and the similarity of study designs. We also provided information on the temporal patterns of adverse events and agree with the recent recommendations that future trial reporting should include such information [7]. Limitations of these findings include those inherent with retrospective, post-hoc analyses, although the data was prospectively collected; no statistical comparisons on safety data were performed. The efficacy of opicapone has been extensively reported in the individual studies [3–5], where it has been shown to significantly reduce OFF time by at least an hour versus placebo (and almost 2 hours versus baseline).
CONCLUSIONS
Long-term use of opicapone once-daily over 1-year at doses of 25 mg or 50 mg was not associated with any new or unexpected safety concern. Across the clinical studies, the percentage of patients who discontinued because of TEAEs was low and similar across the treatment groups. Few patients discontinued opicapone therapy due to TEAEs and the high retention of patients across the double-blind and open-label extension phases support the good tolerability of repeated treatment.
FUNDING
The BIPARK-1 and BIPARK-2 studies were by funded by BIAL - Portela & Ca, S.A.
DECLARATION OF INTERESTS
J.J.F. has held consultancy functions with GlaxoSmithKline, Novartis, TEVA, Lundbeck, Solvay, Abbott, BIAL, Merck-Serono, Merz, Ipsen, Biogen; has received lecture fees from Biogen and BIAL; has received grants from GlaxoSmithKline, Grunenthal, MSD, Allergan, Novartis, Fundação MSD (Portugal) and Teva; has been employed by Centro Hospitalar Lisboa Norte, Faculdade de Medicina de Lisboa.
AL is funded by the Reta Lila Weston Institute of Neurological Studies, University College London, Institute of Neurology and reports consultancies for: Britannia Pharmaceuticals, BIAL Portela. He also reports grants and/or research support: from the PSP Association, Weston Trust, The Reta Lila Howard Foundation and honoraria from Britannia, UCB, Roche, Novartis, Boehringer Ingelheim, Lundbeck, GE Healthcare, Teva, GSK, Ipsen, Allergan, Orion, Bial, AbbVie Lucid, Nordiclnfu Care.
WP reports receiving consulting fees from AbbVie, Allergan, Astra Zeneca, BIA, Boehringer-Ingelheim, Boston Scientific, GlaxoSmithKline, Ipsen, Lundbeck, Medtronic, MSD, Merck-Serono, Merz Pharmaceuticals, Novartis, Orion Pharma, Teva, UCB and Zambon.
OR reports receiving consulting fees from Abbvie, BIAL, Britannia, Lundbeck, Merck, Mundipharma, Sanofi, Servier, Teva, UCB, XénoPort, Zambon; and grant support from Agence Nationale de la Recherche (ANR), Boehringer Ingelheim, CHU de Toulouse, French Parkinson, GSK, INSERM-DHOS, Michael J Fox Foundation, Programme Hospitalier de Recherche Clinique, Recherche Clinique Translationnelle, UCB, Teva and Lundbeck.
JFR, HG and PSS are employed by BIAL – Portela & Co, S.A.
Footnotes
ACKNOWLEDGMENTS
The BIPARK-1 and BIPARK-2 studies were supported and funded by BIAL – Portela & Ca, S.A. Medical writing assistance for this report was provided by Anita Chadha-Patel of ACP Clinical Communications Ltd and was funded by BIAL – Portela & Ca, S.A.