
Abstract
Background
Continuous subcutaneous apomorphine infusion (CSAI) has been used globally since the 1980s for Parkinson disease (PD) motor fluctuations but has not been available in the United States (US).
Objective
Evaluate CSAI for motor fluctuations in the US setting.
Methods
This open-label study (NCT02339064) enrolled patients with PD experiencing ≥3 hours (h) daily OFF time despite optimized levodopa and current/prior use of at least one other adjunctive therapy. CSAI was initiated with a 1–2 mg bolus followed by 1 mg/h infusion titrated to optimal efficacy and tolerability. Following titration, patients entered a 52-week maintenance period.
Results
Of 99 patients treated, 85 completed the titration period, 69 completed maintenance week 12 and 48 completed maintenance week 52. Common treatment-related adverse events included infusion site nodules and erythema, dyskinesia, nausea, and somnolence, each of which occurred more frequently during the titration period. Reduction in OFF time began at CSAI initiation and reached a mean of 3.0 ± 3.18 h/day by maintenance week 12 (primary efficacy endpoint), with a corresponding increase in Good ON time of 3.1 ± 3.35 h/day. By maintenance week 12, 68% of patients rated themselves as much or very much improved, 62% had at least a 2-h reduction in daily OFF time, and mean concomitant oral levodopa and levodopa equivalent doses (excluding CSAI) had been reduced by 198 mg/day and 283 mg/day, respectively. Improvements were maintained through week 52.
Conclusions
This study supports the clinical utility of CSAI to reduce OFF time and increase Good ON time in patients with motor fluctuations inadequately controlled with oral therapy.
Plain language summary
Continuous subcutaneous apomorphine infusion (also called CSAI) is a treatment to control OFF time (periods of poor motor control) that occurs despite ongoing use of usual Parkinson disease medications like levodopa. Apomorphine is slowly infused under the skin using a thin, flexible tube connected to a small, wearable infusion device. CSAI therapy has been widely used outside the United States for its effective control of Parkinson disease symptoms since the mid 1980s. In order to gain approval for CSAI use in the United States, a specific study conducted in United States clinics and hospitals was required. In this study, people with Parkinson disease experiencing 3 or more hours of OFF time per day were started on apomorphine infusion (CSAI) therapy. The dose of apomorphine was increased until each study participant experienced the best possible benefit while still tolerating the medication. Once this dose was identified, participants continued on this dose. The most common side effects were nodules (small bumps under the skin) and erythema (redness of the skin) at the infusion site, dyskinesia (involuntary movements), nausea, and somnolence (sleepiness). These side effects were more frequently experienced at the start of treatment when the best dose of CSAI for each participant was being identified and the participant's other PD medications were being adjusted. Soon after they started CSAI treatment, participants noticed a reduction in the amount of OFF time that they experienced each day. After 12 weeks at a stable dose of CSAI treatment, participants' OFF time had decreased by about 3 hours per day on average. At the same time, most (68%) participants said they felt their symptom control was “much improved“ or “very much improved” compared to before starting CSAI therapy. Importantly, these improvements were maintained throughout one year of treatment. These findings support the use of apomorphine infusion in the United States as an effective and safe treatment option for people with Parkinson disease that does not require surgery.
Introduction
Apomorphine has receptor affinity and structural similarity to dopamine. Like endogenous dopamine, apomorphine activates all dopamine receptor subtypes and is effective at reliably producing an ON state. The antiparkinsonian effect of apomorphine is thought to result from activation of both D1-family (D1 and D5) and D2-family (D2, D3, D4) dopamine receptors. Unlike conventional dopamine receptor agonists that are D2-family predominant, this dopamine-like dual activation of both D1- and D2-family receptors is thought to underlie both apomorphine's robust efficacy and also its lower incidence of adverse effects associated with D2-family predominant receptor agonists (such as impulse control disorders). 1
Apomorphine has been long used as a treatment for Parkinson disease (PD).2–4 Although approved in the US in 2004 as a subcutaneous injection for on-demand, acute treatment of OFF episodes, 5 it has not been available in the US as a continuous subcutaneous apomorphine infusion (CSAI) to treat recurrent motor fluctuations. Widely used for decades in Europe, Australia, and Asia, CSAI was studied in a randomized, double-blind, placebo controlled European trial (TOLEDO) in 2018. 6 TOLEDO provided level I evidence for efficacy and safety of CSAI in treating patients with recurrent motor fluctuations despite optimized levodopa with adjunctive medication. In the TOLEDO study, CSAI demonstrated good tolerability and significantly reduced daily OFF time versus placebo, with 62% of patients achieving a reduction of ≥2 h of OFF time per day. 6 For patients who continued into the TOLEDO extension study, the efficacy of CSAI was sustained for 52 weeks (mean daily OFF time was reduced by 3.7 h from double-blind baseline, equating to a 53% reduction). 7 While the pivotal TOLEDO trial was ongoing in Europe, we initiated a prospective, long-term, open-label, phase 3 clinical trial (InfusON) in the US to evaluate the safety, tolerability, and efficacy of CSAI in PD outpatients with motor fluctuations despite optimized treatment with available PD therapies, including oral levodopa and current or prior use of at least one other PD therapy. The InfusON trial is the first, and to date, the only study of CSAI in the US.
Methods
Study conduct
This was a prospective, 52-week open-label, phase 3, outpatient study conducted at 19 US centers with expertise at treating motor fluctuations in PD. The study was registered on www.clinicaltrials.gov as NCT02339064, and conducted in accordance with the Declaration of Helsinki, Good Clinical Practice guidelines and US Code of Federal Regulations dealing with Protection of Human Patients (US 21 CFR Part 50). The study protocol was reviewed and approved by central or local institutional review boards for each site. Written informed consent was obtained from all patients and from care partners involved in assisting patients.
Study population
We recruited levodopa-treated PD patients (>30 years of age) whose control of motor fluctuations was judged unsatisfactory by both the investigator and the patient despite prior attempts with oral carbidopa/levodopa adjustments and adjunctive therapies to optimize treatment response. Enrolled patients experienced ≥3 h of daily OFF time, despite a stable regimen of oral carbidopa/levodopa and at least one other current or prior adjunctive therapy (i.e., oral or transdermal dopamine receptor agonist, monoamine oxidase type-B inhibitor, catechol-O-methyltransferase inhibitor, amantadine, deep brain stimulation [DBS], or levodopa/carbidopa intestinal gel infusion). Patients using intermittent apomorphine subcutaneous injections were allowed to continue to use these injections as needed in addition to CSAI. Patients who needed help with study procedures (including device use and motor diary keeping) were required to have a care partner able to assist throughout the study.
Main exclusion criteria were: use of 5HT3 receptor antagonists (e.g., ondansetron), nitrates, or antidopaminergic drugs, recent history of impulse control disorder (ICD) or of alcohol or substance abuse, clinically significant renal disease (estimated glomerular filtration rate <60 mL/min/1.73 m2), clinically significant orthostatic hypotension, history of QT interval prolongation or current corrected QT interval (QTcF) ≥ 450 ms, heart rate <55 or >100 beats per minute (bpm), systolic blood pressure <90 mmHg, atrial fibrillation with a poorly controlled rate, recent (past year) myocardial infarction, history of malignant melanoma, planned DBS for PD, clinically significant cognitive impairment or any other condition that might interfere with study procedure compliance.
Study design
The study consisted of screening (including a baseline period), titration, and maintenance periods. An extension period (ongoing) was also added to allow patients to remain on CSAI treatment until commercial availability (to be reported separately).
During the screening period (up to 35 days prior to CSAI initiation), patients and care partners received training in diary assessments (including recognition of motor states for Hauser diary evaluations 8 ), care and operation of the ambulatory infusion device, infusion technique, and infusion site care, including the need to change the infusion site daily. Upon completion of training, patients entered the baseline period, which began at least 14 days prior to CSAI initiation and was contained within the 35-day screening window. During the baseline period, the investigator confirmed that the patient's PD regimen was optimized (i.e., no further benefit would result from PD medication changes); the patient's motor fluctuations persisted despite optimized therapy; and the patient was able to complete motor diaries. During the baseline period the patient completed two sets of 3-day motor diary recordings: the first set (practice set) was completed during the first 3 days of the baseline period (with telephone follow-up on the fourth day); the second set was completed immediately prior to the CSAI initiation visit and was used to confirm eligibility requirements and serve as the baseline measure for efficacy assessments.
Apomorphine for CSAI was provided in 20 mL pre-filled glass cartridges and delivered via a wearable (ambulatory) infusion device. Each mL of solution contained 5 mg apomorphine hydrochloride hemihydrate (equivalent to 4.27 mg apomorphine base). Participants whose daily infusion needs totaled 100 mg (20 mL) or more, including bolus doses and solution for priming, required more than one cartridge per day. For this report, infusion rates are described in terms of mg of apomorphine, meaning mg of apomorphine HCl hemihydrate, which is consistent with the way dosing is expressed in the TOLEDO study. 6
Patients initiated CSAI in the outpatient setting at least 3 h after the last levodopa dose, with a 2 mg bolus dose (1 mg for patients with renal impairment) and an initial infusion rate of 1 mg (0.2 mL) per hour. Patients received trimethobenzamide (Tigan) 300 mg, three times daily, starting 2–3 days prior to CSAI initiation (optional for apomorphine injection users) with continued use of trimethobenzamide encouraged through week 12, though use could be discontinued at the investigator's discretion.
During the titration period, the CSAI rate was individually optimized to achieve best waking-day efficacy without intolerable adverse effects (AEs). The infusion rate was adjusted over the next 3–5 visits (in 0.5–1.0 mg/h increments) according to clinical response and tolerability to a maximum of 8 mg/h and allowed up to 3 daily bolus doses of 1–2 mg. The total daily apomorphine dose was not to exceed 150 mg/day. Titration visits occurred 1–10 days apart. Patients were to maintain daily infusion during waking hours, with a target of 16 h/day. Use of the infusion up to 24 h/day was permitted at the investigator's discretion. After the device was programed by trial site staff, settings were locked at identified flow rate and bolus dose settings.
No increase or addition to baseline PD medications was allowed for motor control during titration. Protocol recommendations to manage treatment-emergent AEs, including dyskinesia, were that the investigator should first reduce oral/transdermal dopamine receptor agonists, and then reduce other baseline PD medications. If CSAI needed to be reduced (for an AE that could not be managed by reduction of baseline PD medications), then bolus doses were to be reduced (or discontinued) first, followed by infusion rate reductions in 0.5–1.0 mg/h increments. The minimum required infusion rate to enter the maintenance period was 1 mg/h (0.2 ml/h); patients who did not tolerate this infusion rate were discontinued from the study.
Patients entered the maintenance period once their optimized CSAI and PD medication dosage regimen was identified during the titration period. For the first 12 weeks of the maintenance period, the CSAI rate and PD medications were to remain unchanged. After 12 weeks, the protocol allowed incremental CSAI adjustments (not exceeding 8 mg/h, or 150 mg/day). Patients who withdrew after the week 12 maintenance period visit had the CSAI tapered slowly over 8 days before discontinuing treatment. Upon completing the 52-week maintenance period, patients could enter the extension period, which is ongoing.
Outcome measures
Consistent with the primary objective, safety and tolerability were determined through adverse event (AE) reporting, clinical monitoring (laboratory test results, vital signs, and ECGs), physical and dermatologic examinations, and rating scales (Epworth Sleepiness Scale (ESS), 9 the Questionnaire for Impulsive-Compulsive Disorders in Parkinson's Disease Rating Scale (QUIP-RS), 10 and the Columbia Suicide Severity Rating Scale (C-SSRS) 11 ). Additionally, three AEs of special interest (AESI) were prospectively identified for monitoring: 1) infusion site reactions (ISRs) that prevented re-use of the same site for drug delivery for more than 5 days, or caused pruritis severe enough to require treatment, or showed signs of infection; 2) symptomatic orthostatic hypotension (defined as a drop in systolic pressure ≥20 mmHg or in diastolic pressure ≥10 mmHg that was accompanied by lightheadedness or dizziness), and 3) symptoms or signs of potential hemolytic anemia, including a hemoglobin drop ≥1.5 g/dL compared to screening or prior visit. The presence of laboratory-confirmed hemolytic anemia required discontinuation from the study.
The primary efficacy endpoint was change from baseline to week 12 of the maintenance period in total daily OFF time, as recorded in the 24-h motor-diaries.
8
Patients recorded their predominant motor state in 30-min increments, and entries for time sleeping were recorded upon awakening. Key secondary efficacy measures were the change from baseline to week 12 of the maintenance period in:
Daily ON time without troublesome dyskinesia (commonly referred to as Good ON time
12
) Patient Global Impression of Change (PGI-C)
13
Oral levodopa and levodopa equivalent dose (LED)
14
Percentage of patients with response to therapy, defined as OFF time reduction of ≥2 h/day.
Other secondary measures (completed at baseline, weeks 2 and 12 of the maintenance period and every 8 weeks thereafter) included the Unified Parkinson's Disease Rating Scale (UPDRS) Parts II (activities of daily living) and III (motor exam),
15
Clinical Global Impression of Change (CGI-C), and the Parkinson's Disease Questionnaire (PDQ-39).
16
The Non-Motor Symptoms Scale (NMSS),
17
was assessed as an exploratory outcome.
Statistical analysis
No sample size estimation was performed as this trial was primarily designed as a safety study and conducted using an open-label design. A planned enrollment of up to 100 patients was considered to be adequate for meeting study objectives. Safety was assessed for all patients who received ≥1 dose of study medication (Safety Set) and efficacy was assessed for the modified intent to treat (mITT) population, which included all Safety Set patients who had a valid assessment of OFF time at both baseline and any post-baseline visit. For diary analyses, daily time in each motor state was calculated by taking the average of valid diaries recorded for the 3 days before the corresponding clinic visit. Diary recordings were considered valid if there were ≤4 missing 30-min motor score entries per 24 h. In addition, an analysis of motor states was done, to evaluate the percentage of OFF and ON time experienced during the time the device was in use.
Results
Study population
Between 31 March 2014 and 26 July 2018, 153 patients were screened, of whom 99 were enrolled and initiated CSAI treatment (Figure 1). Dose initiation was generally well tolerated with 4 patients discontinuing treatment during week 1 (n = 2 at the initiation visit, n = 1 on day 3 and n = 1 on day 5), and one additional patient discontinuing during week 2, while still on the initiation dose; none of these 5 patients had post-baseline efficacy data, leaving 94 patients in the mITT population. Overall, 85 patients completed the titration period, 69 completed the first 12 weeks of the maintenance period, 65 completed at least 6 months of treatment, and 48 completed 52 weeks. Most (n = 45) patients who completed the 12-month maintenance period elected to continue CSAI in the ongoing extension period.
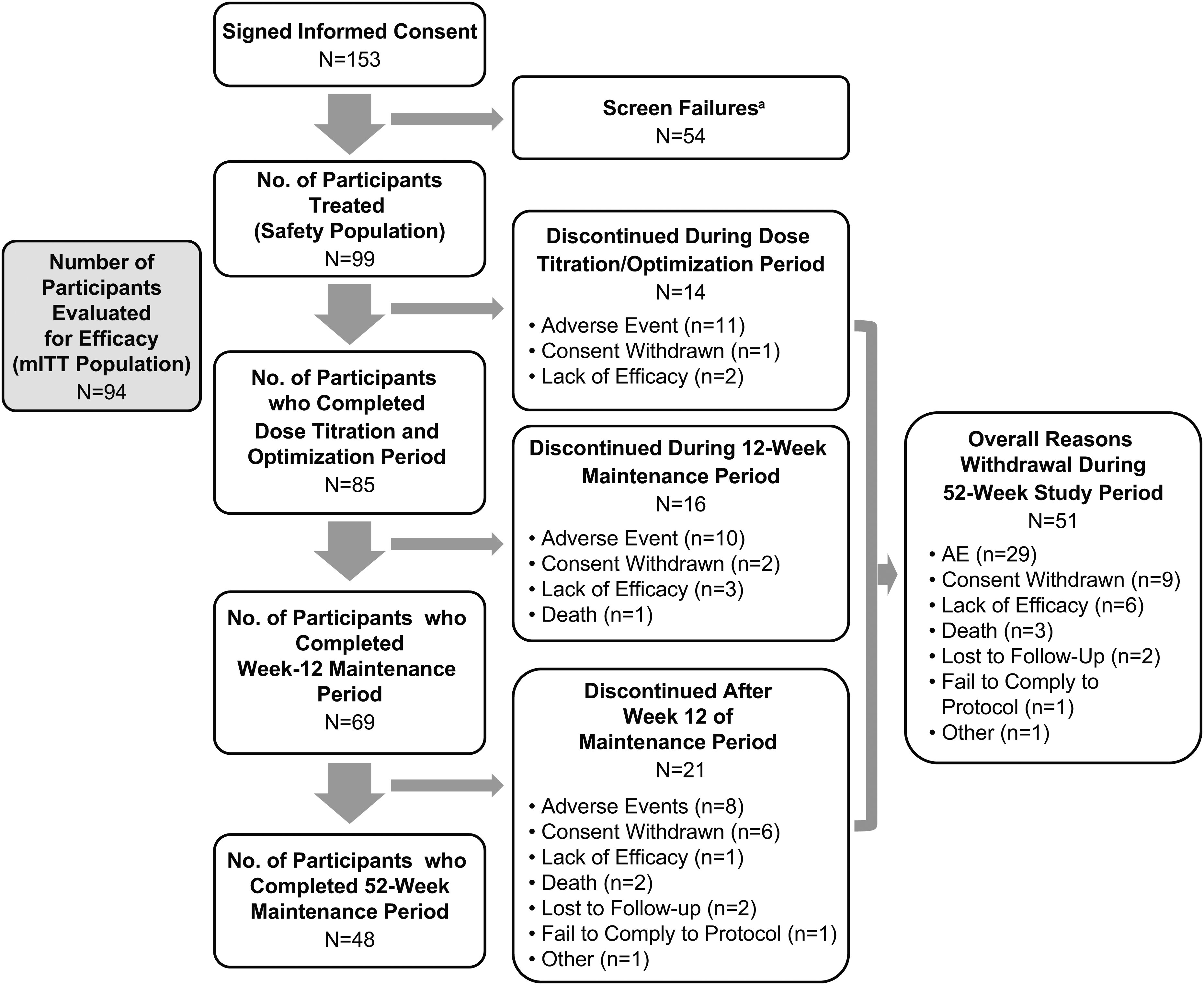
Study disposition. aReasons for screen failure (participants may have more than one): not able/willing to comply with scheduled visits, treatment plan, study procedures (n = 17); not able to correctly recognize “ON” “OFF” and “dyskinesia” states, or maintain motor diary or infusion log (n = 6); did not have unsatisfactory motor control once treatment was optimized (n = 6); presence/history of QTc prolongation, heart rate <55 bpm or >100 bpm or clinically significant orthostasis (n = 6); OFF periods <3 h/day during baseline (n = 5); no caregiver available, or caregiver unable/unwilling to consistently assist subject (n = 5); presence ≥2 h/day troublesome dyskinesia (n = 4); history or presence of ICD associated with dopamine receptor agonists (n = 4); significant cognitive impairment or surgical, neurologic, psychiatric, or laboratory condition that would interfere with participation (n = 4); participant/caregiver unable to master use of CSAI delivery system (n = 2); history/current malignant melanoma (n = 2); did not have advanced PD consistent with UK Brain Bank Criteria (n = 1); planned surgical treatment for PD (n = 1); clinically significant renal disease (GFR <60 mL/min/1.73 m2 [n = 1]); known/suspected pregnancy or currently lactating (n = 1); reason not documented (n = 1). mITT: modified intent to treat.
Baseline demographics are shown in Table 1. Patients were predominantly male (69.7%), the mean age was 61.6 years; duration of PD was 10.0 years; duration of PD-related motor fluctuations was 5.3 years, and mean daily OFF time was 6.6 h. Mean daily ON time with troublesome dyskinesia was 0.5 h.
Baseline characteristics.

Hoehn & Yahr stage was to be assessed in the OFF state, ≥ 3 h after the last levodopa dose. bModified intent to treat population.
PD medications and CSAI throughout the study
At baseline, all patients were taking levodopa (mean dose: 1063 mg/day [Safety Set, n = 99]), with 90.9% (n = 90) also concomitantly using other adjunctive PD therapies, most commonly dopamine receptor agonists (79.8%) (Table 2). Of the 15 patients (15.2%) who had been using intermittent apomorphine injections (Apokyn) prior to baseline, 9 (9.1%) continued use during the trial, 5 (5.1%) discontinued use within one day of starting CSAI, and 1 (1.0%) discontinued use ∼1 month before study start. Additionally, of 8 (8.1%) patients with a history of DBS placement, 5 (5.1%) continued DBS use during the trial and 3 had discontinued use at some point prior to study treatment. Most patients (n = 89, 89.9%) were administered trimethobenzamide prior to CSAI initiation, 9 (9.1%) did not use trimethobenzamide at all, and 1 (1.0%) patient had unconfirmed use. Of the 89 using trimethobenzamide, 18 (18.2%) used it throughout their study participation, and 71 (71.7%) discontinued use, with most (58 [58.6%]) doing so during the titration or early maintenance period (maintenance weeks 1–12).
Antiparkinsonian medication use and dosing parameters for CSAI by study period.
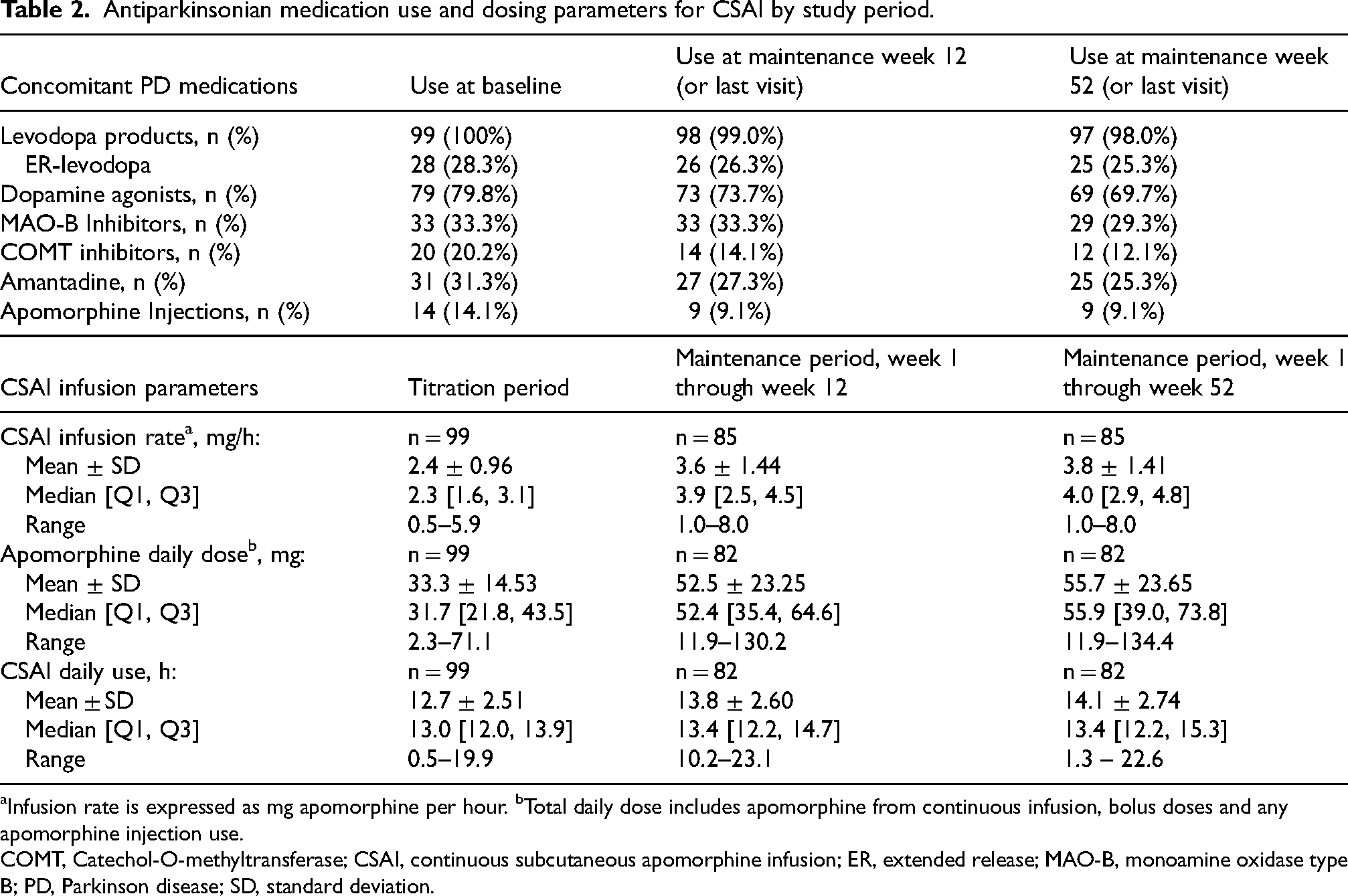
aInfusion rate is expressed as mg apomorphine per hour. bTotal daily dose includes apomorphine from continuous infusion, bolus doses and any apomorphine injection use.
COMT, Catechol-O-methyltransferase; CSAI, continuous subcutaneous apomorphine infusion; ER, extended release; MAO-B, monoamine oxidase type B; PD, Parkinson disease; SD, standard deviation.
The median duration of CSAI dose titration was 30 days and patients typically required 3 to 5 (median = 4 and mode = 4) infusion dose adjustments to reach their optimal maintenance dose. Dosing parameters (CSAI and other PD medications) are given in Table 2 and the distribution of patient CSAI maintenance doses and infusion rates are shown in Supplemental Figure 1. Most patients were maintained on an infusion rate of 3 to 5 mg/h, used the device between 12 and 16 h/day, and received between 40 mg and 80 mg of apomorphine per day (including bolus and/or apomorphine injection doses). A subset of 13 (13.1%) patients opted for overnight infusions (i.e., up to 24 h per day); 84.6% (n = 11) of these patients completed the 52-week maintenance period.
Safety
All patients reported one or more AEs (Table 3, Supplemental Table 1), which were mostly mild to moderate in severity. The most commonly reported AEs considered at least possibly related to CSAI treatment were infusion site nodules, dyskinesia (all patients had dyskinesia or history of dyskinesia at baseline), infusion site erythema, nausea, somnolence, and dizziness, all of which were reported with higher incidence during the titration period relative to the maintenance period. Ten patients (10.1%) experienced one or more serious AEs (SAE), all during maintenance therapy. Three of these 10 patients (3.0% of total patients) had an SAE that was considered potentially treatment related by the investigator: one patient had an SAE of angina pectoris (day 251), one had an SAE of dyskinesia (day 118), and one had an SAE of paranoia (day 266). Of these 3 events, only the AE of paranoia led to treatment discontinuation (on day 313). Three patients (3.0%) died during the 52-week maintenance period: one resulting from a traffic accident (day 168), one resulting from brain hypoxia and aspiration (day 352) and one resulting from aspiration pneumonia (day 131); none were considered treatment related.
Adverse Events.a
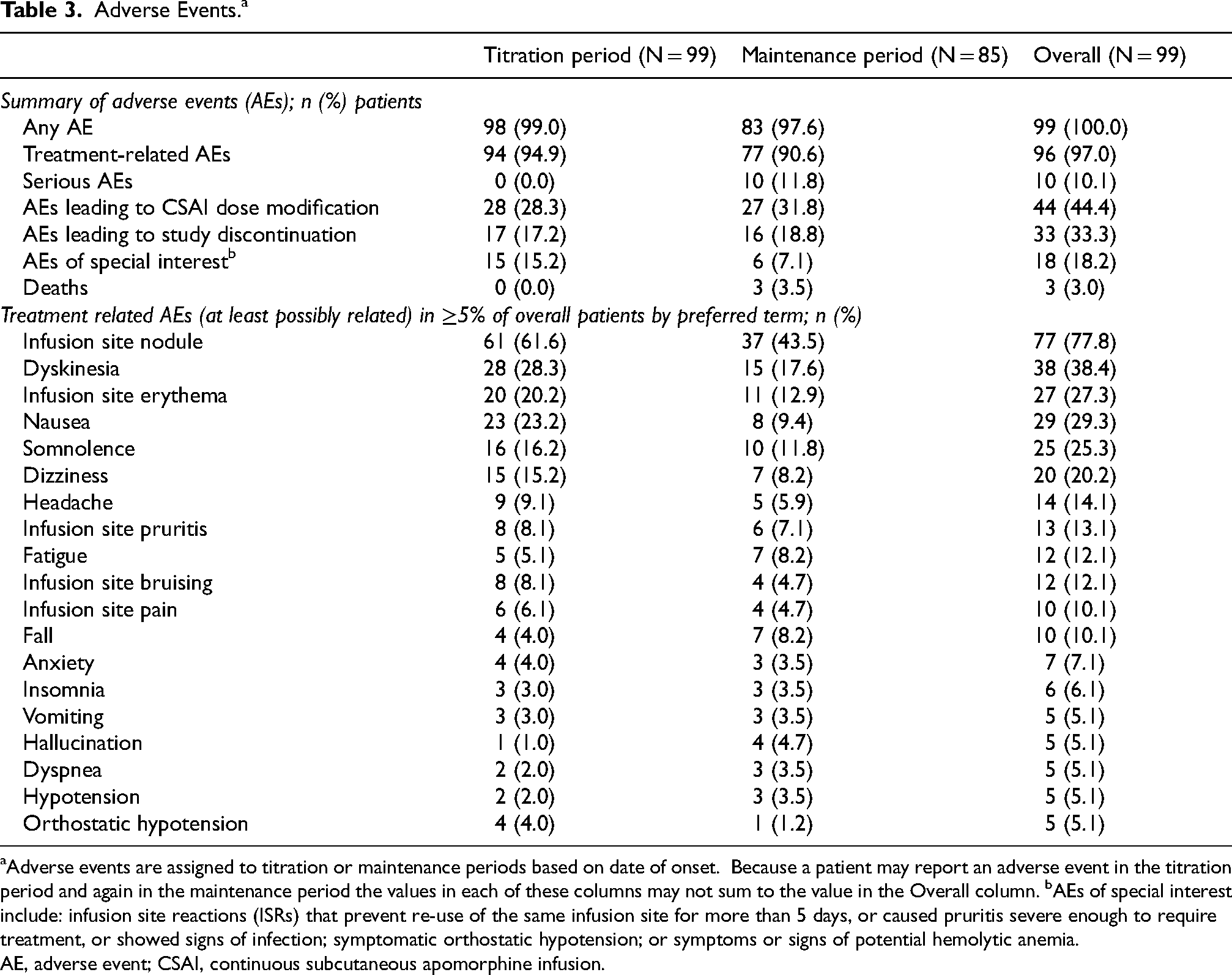
aAdverse events are assigned to titration or maintenance periods based on date of onset. Because a patient may report an adverse event in the titration period and again in the maintenance period the values in each of these columns may not sum to the value in the Overall column. bAEs of special interest include: infusion site reactions (ISRs) that prevent re-use of the same infusion site for more than 5 days, or caused pruritis severe enough to require treatment, or showed signs of infection; symptomatic orthostatic hypotension; or symptoms or signs of potential hemolytic anemia.
AE, adverse event; CSAI, continuous subcutaneous apomorphine infusion.
Thirty-three patients (33.3%) had AEs leading to treatment discontinuation. For approximately half (n = 17), these AEs were first reported during the titration period. The most common AEs leading to discontinuation were infusion site nodules (n = 5 [5.1%]; n = 4 during titration), fatigue and dizziness (n = 4 [4.0%] each, 2 each during titration) and nausea (n = 3 [3.0%], all during titration). In addition to the 5 patients who discontinued for skin nodules (one of whom also reported mild infusion site pain and erythema), 3 patients discontinued for other infusions site conditions, including hypersensitivity [n = 1], pain at infusion site [n = 1], and urticaria [n = 1]; all of these were mild or moderate in severity. Forty-four patients (44.4%) had one or more AEs leading to CSAI dose modification (increase, decrease, or dose interruption), most commonly, dyskinesia (9.1%), somnolence (6.1%), anxiety (5.1%) and dizziness (5.1%).
AESI were reported for 18 patients (18.2%). The majority of AESI were infusion-site related, including infusion site pruritus requiring symptomatic treatment (n = 8 [8.1%]) and infusion site skin reactions that either prevented re-use of the infusion site for ≥5 days (n = 5 [5.1%]), or showed signs of infection (n = 2 [2%]). Symptomatic orthostatic hypotension with dizziness or lightheadedness was experienced by 5 patients (5.1%), including 4 [4%] during titration and 1 [1.2%] during maintenance. Hemolytic anemia was not reported, and although decreases in hemoglobin and/or positive Coomb's tests occurred during the study (Supplemental Table 2), these did not result in treatment discontinuation. Infusion device events were reported for 12 patients (12.1%), including device malfunction, e.g., display screen or battery issues (7 patients [7.1%]) and catheter tip breakage (5 patients [5.1%]).
Mean total ESS and QUIP-RS scores were within normal ranges at baseline and all study visits (Supplemental Table 3). Fifteen patients (15.2%) reported any new instances of unintended or unwanted falling asleep in undesirable situations at any post-baseline visit. Of these 15 patients, 13 (86.7%) were using concomitant dopamine receptor agonists (similar to the overall sample), and “sleep attack” was reported as an AE for only 2 of the patients (both of whom were using dopamine agonists). Eleven patients reported adverse events potentially suggestive of impulse control disorders including compulsivity (gambling, sexuality, shopping, or OCD); of these 11 patients, 9 (81.8%) were using concomitant dopamine receptor agonists (again, similar to the overall sample). Seven patients (7.1%) reported suicidal ideation on the C-SSRS during the 52-week study, which was the same as the total number reporting suicidal ideation or history of suicidal ideation on the baseline assessment. No patient reported suicidal behavior during the 52-week study (2 reported past suicidal attempts at baseline).
Evaluation of laboratory test values did not show meaningful trends. Abnormal blood pressure and heart rate recordings were somewhat more common during titration than maintenance period visits. Increases or decreases >25% in supine-to-standing blood pressure (systolic or diastolic) occurred in 32.3% and 35.4% of patients, respectively, at one or more study visits. Increases or decreases >25% in supine-to-standing heart rate occurred in 56.6% and 8.1% of patients, respectively, at one or more visits. Three patients (3.0%) experienced ECG-related AEs (electrocardiogram QT prolonged); all were considered mild in severity; 2 of these 3 were discontinued (per protocol) during the titration period and one continued in the study following an apomorphine dose reduction.
Efficacy
At baseline, the mean ± SD daily OFF time was 6.6 ± 2.36 h/day (n = 94). Improvement was observed already on the initiation dose, with diaries collected at the first return clinic visit (Visit 2) showing a change in OFF time of −0.9 ± 3.18 h/day (n = 82). Improvement (reductions) in OFF time continued at subsequent visits, reaching −1.7 ± 2.81 h/day (n = 92) on diaries collected at the second return clinic visit, and −2.8 ± 2.88 h/day on those returned at end of titration (maintenance day 1), at which time the mean ± SD daily OFF time was 3.8 ± 2.53 h/day (n = 80). Reduction in OFF time was sustained throughout the maintenance period, with mean changes from baseline of −3.0 ± 3.18 h/day at maintenance week 12 (primary endpoint) and −3.2 ± 3.23 h/day (n = 48) at maintenance week 52 (Figure 2).
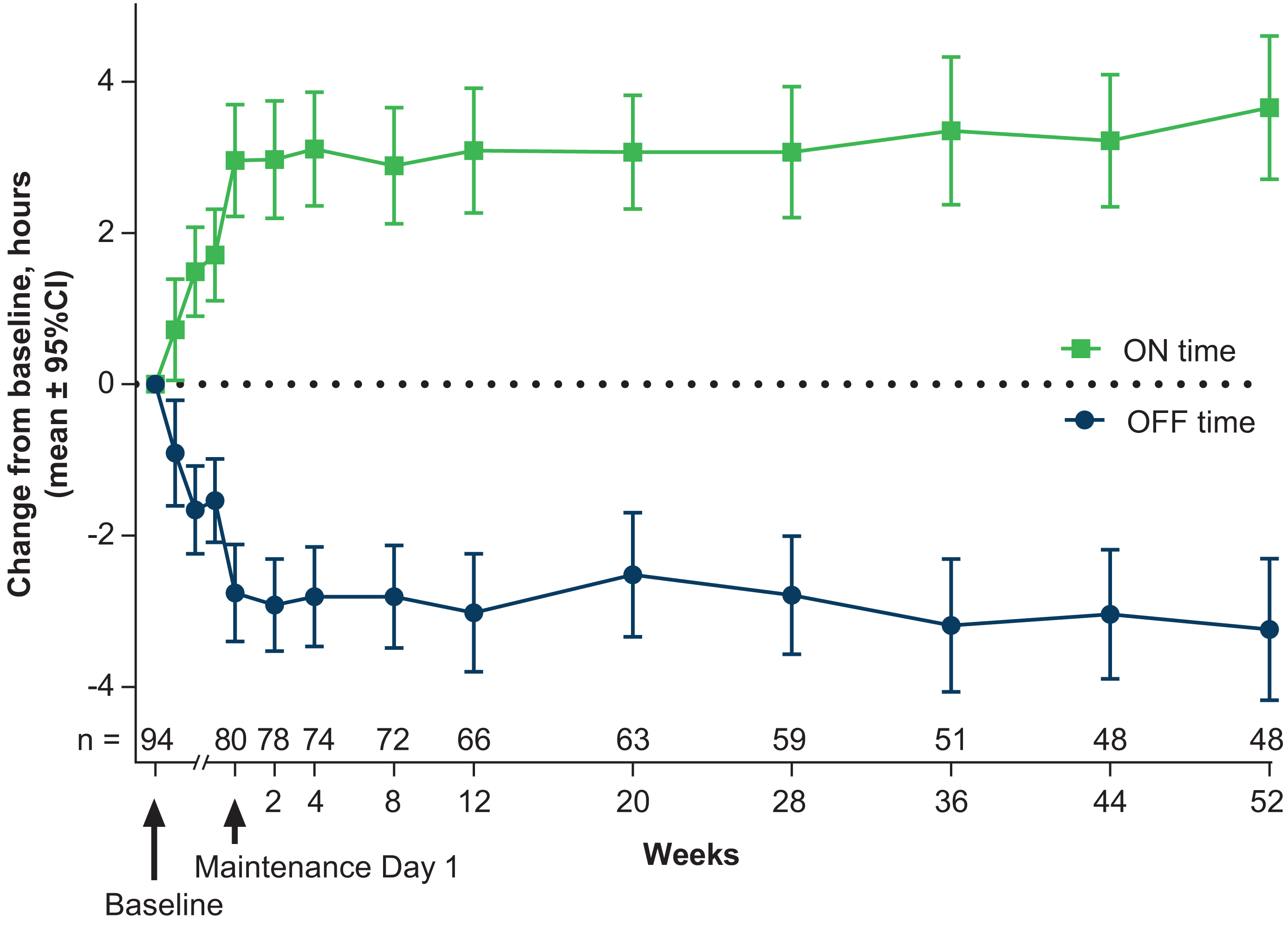
Mean changes from baseline in daily OFF time and daily ON time without troublesome dyskinesia (mITT population). The blue line shows reduction from baseline in daily OFF time (primary efficacy outcome). The green line shows improvement from baseline in daily ON time without troublesome dyskinesia (also called Good ON time; key secondary outcome). The median length of the titration period (between baseline and maintenance day 1) was 30 days. The 3 data points prior to maintenance day 1 represent scheduled titration visits following treatment initiation. Sample sizes for these data points are n = 82, n = 92 and n = 90, respectfully. Data for any optional titration visits are not graphed. Labels for Weeks on the x-axis represent weeks following the start of maintenance treatment.
Reductions from baseline in daily OFF time were mirrored by incremental improvements in Good ON time that were apparent on the initiation dose (+0.7 ± 3.06 h/day on diaries returned at Visit 2), and sustained throughout the maintenance period, with changes (increases) on maintenance day 1 of +3.0 ± 3.33 h/day, maintenance week 12 of +3.1 ± 3.35 h/day (first key secondary outcome), and maintenance week 52 of +3.7 ± 3.28 h/day. Small reductions in ON time with troublesome dyskinesia were observed at all maintenance visits (e.g., 0.28 [1.62] h/day at maintenance week 12).
The responder analysis showed 26.8% of patients experienced an OFF-time reduction ≥2 h/day by the first return titration visit (while still on the initiation dose). The responder rate increased to 61.3% by end of titration (maintenance day 1) and remained stable thereafter with 62.1% meeting responder criteria at maintenance week 12 (key secondary outcome) and 64.6% at maintenance week 52 (Table 4). Additional analysis confirmed that the improvements in OFF time and ON time without troublesome dyskinesia were largely seen during the time the CSAI device was being used (Supplemental Figure 2).
Efficacy measures.

UPDRS scores assessed at least 3 h post-levodopa dose. The sample sizes for each assessment are shown in column1 of the Table and were used as denominators for any percentage calculations.
NMSS, Non-Motor Symptoms Scale; PDQ-39, Parkinson's Disease Questionnaire, 39 items; UPDRS, Unified Parkinson's Disease Rating Scale.
Of the 5 patients who used DBS during the study, 4 completed maintenance week 12; one patient discontinued on day 32 for lack of efficacy, one discontinued due to the SAE of paranoia (Study day 313), and 3 completed and continued treatment in the study extension period.
By maintenance week 12, 67 of the 75 patients (89.3%) who completed the PGI-C (key secondary endpoint) reported improvement, with 51 (68%) rating themselves “much improved” or “very much improved” (Figure 3) and the proportion reporting improvement was maintained through maintenance week 52. This patient-reported outcome mirrored clinician-rated improvement on CGI-C (Figure 3).
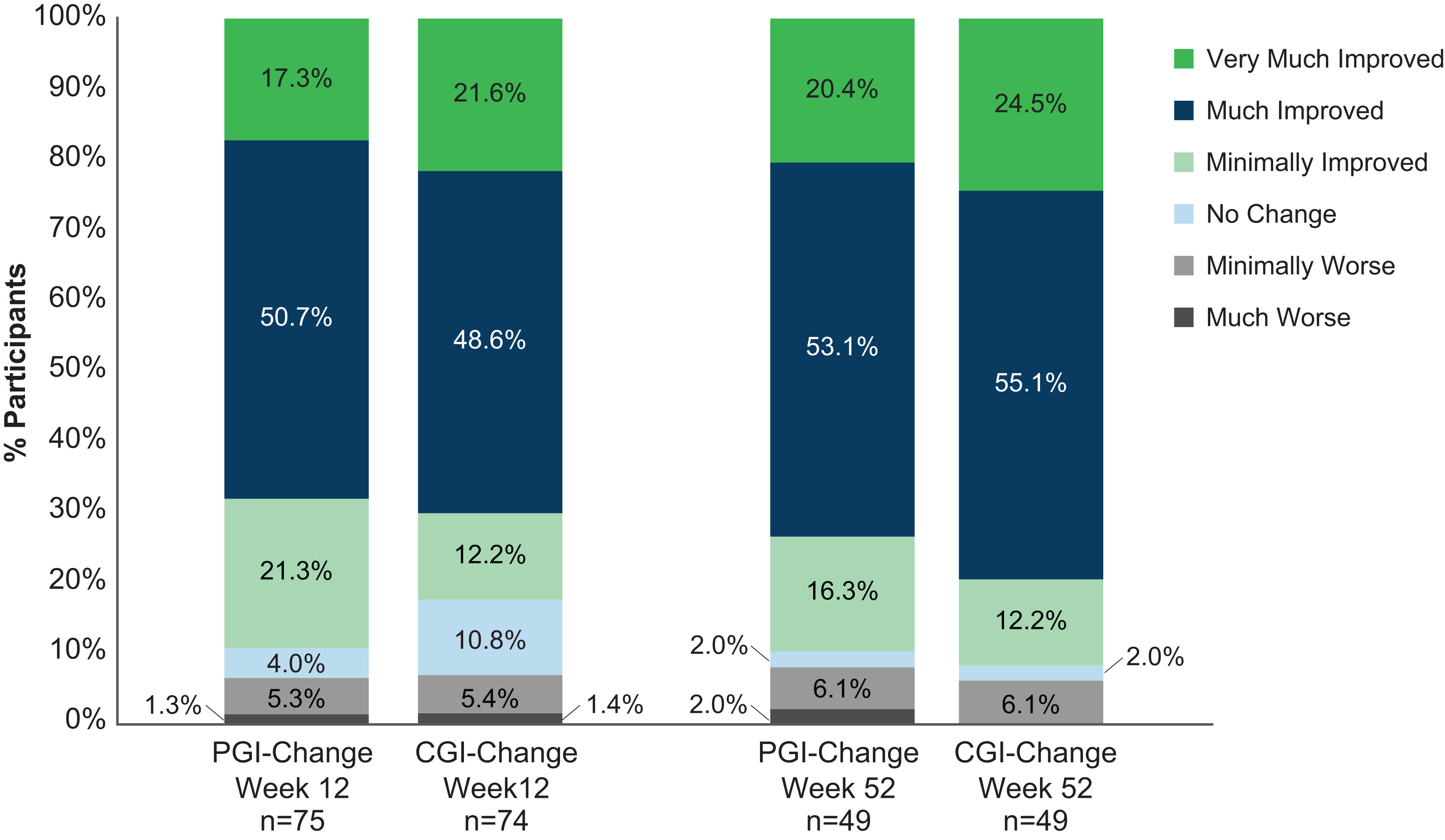
Patient Global Impression of Change (PGI-C) and Clinical Global Impression of Change (CGI-C) at maintenance period week 12 and week 52.
In this trial, treatment emergent dopaminergic AEs, such as dyskinesia, were to be managed by lowering baseline adjunctive PD medications, first oral/transdermal dopamine receptor agonists, then other adjunctive medications and carbidopa/levodopa. Overall, 48 patients (48.5%) reduced the dose of one or more adjunctive PD medications during the titration period and 24 patients (24.2%) had completely eliminated ≥1 class of these medications by week 52 or last study visit (2 patients had added a medication class by week 52 or last visit).
Daily oral levodopa dose and LED (without apomorphine) were reduced by −161 ± 315.5 mg/day and −239 ± 414.6 mg/day, respectively, by the end of titration (maintenance day 1) and remained below baseline at maintenance weeks 12 (−198 ± 384.8 mg/day and −283 ± 504.1 mg/day [key secondary outcomes]) and 52 (−128 ± 593.4 mg/day and −250 ± 654.2 mg/day), with decreases in levodopa use accounting for half or more of the LED reductions. Consistent with CSAI use as adjunctive therapy, adding infused apomorphine to the calculation (LED with apomorphine) resulted in a net increase in LED from baseline of 252 ± 384.9 mg/day, 245 ± 530.9 mg/day, and 323 ± 627.3 mg/day, at maintenance day 1, week 12, and week 52, respectively.
Improvements were also observed at week 12 for UPDRS Part II (ADL) and Part III (motor) scores (Table 4). Reductions (improvement) in PDQ-39 total and NMSS total scores were also observed, starting at the first assessment of these measures at the maintenance week 2 assessment (Table 4). Improvements on PDQ-39 total scores were seen throughout the study and were predominantly driven by individual PDQ-39 domains of (weeks 12 and 52, respectively) mobility (−9.5 and −8.8), ADL (−6.5 and −6.5), stigma (−8.2 and −8.0), emotional well-being (−3.9 and −2.3), and bodily discomfort (−5.6 and −5.4) (Supplemental Table 4). NMSS total score improvement peaked by week 20 (peak improvement: −6.6 points); howver, small but steady improvement was observed throughout 52 weeks of treatment in the domains of “mood/cognition” and “miscellaneous” (Supplemental Table 5).
Discussion
This phase 3 study provides the first prospective, long-term safety and efficacy data for CSAI in the US. Treatment of motor fluctuations with CSAI demonstrated safety, tolerability, and reduced daily OFF time by over 3 h/day with corresponding increases in Good ON time. Clinical improvement increased through titration, remained stable during the initial 12-weeks of the maintenance period. This improvement persisted through maintenance week 52. Most adverse events were mild or moderate in intensity and did not lead to treatment discontinuation.
Prospective monitoring of safety data for up to 52 weeks in this US patient cohort confirmed that CSAI was generally well tolerated, with a profile of treatment-related AEs consistent with the European TOLEDO studies6,7 and prior open-label apomorphine infusion studies outside of the US. 18 Rates of AEs and early drop-out due to AEs were, however, somewhat higher during initial treatment in our study, where 11 [11.1%] patients discontinued due to AEs during dose optimization vs. 2 [3.7%] in the apomorphine arm of the double-blind TOLEDO study. Reasons for this difference (InfusON vs. TOLEDO) require further exploration but may include less familiarity of the US investigators with subcutaneous therapy (vs. the long-standing experience of European investigators) as well as differences in the titration setting (outpatient vs. inpatient), antiemetic use (trimethobenzamide vs. domperidone), and the comparatively smaller reduction in concomitant PD medication doses (LED reduction of −283 mg by maintenance week 12 vs. −492 mg in TOLEDO). 6
Most patients experienced one or more ISRs; however, these were mild-to-moderate in intensity, infrequently led to study discontinuation (8% of patients) and did not impact efficacy. ISRs are not uncommon in CSAI treated patients, and recent biopsy studies suggest these may involve local inflammatory reactions in subcutaneous tissue.19,20 Additionally, clinical experience in Europe suggests practical aspects such as infusion set selection are also important. For example, previous use of butterfly infusion sets, with stainless steel needles that had to be inserted at a 45° angle, were less comfortable for patients, more difficult to place appropriately, and posed greater problems with skin tolerability. Newer infusion sets, that use integrated insertion devices containing fine-gauge, 90-degree introducer needles to place soft cannulas (such as the Cleo™ sets used in this study, and the Neria™Guard infusion sets widely used in Europe) have been shown to provide better skin tolerability and improved patient experience.21–23 Most CSAI-related ISRs are self-limited, resolve spontaneously, and do not limit successful continuation of CSAI treatment. Clinical guidelines for evaluating and managing ISRs during CSAI therapy have been proposed.21,24
Reductions in OFF time in this US study are highly consistent with both TOLEDO and with other open-label studies (including the TOLEDO open-label extension).7,18,21 The secondary outcome measures also support the efficacy of CSAI, with most patients reporting feeling ‘much- or very much improved’ on PGI-C, despite the fact their baseline PD medications were previously optimized prior to trial entry. Quality of life improvements (PDQ-39) were mainly driven by the individual domains expected to be influenced by better control of motor symptoms (ON and OFF time) including mobility, ADL, and bodily discomfort. Consistent improvements were also seen using the UPDRS Part II (Activities of Daily Living). Changes in non-motor symptoms of PD, as assessed by the NMSS, showed initial improvement followed by return towards baseline status over the 52-week maintenance period; improvements were largely driven by changes in mood and cognition, sleep disturbance, and fatigue components.
In this study, trimethobenzamide was used to try to reduce incidence of nausea despite paucity of evidence to support its antiemetic efficacy. 25 Trimethobenzamide is no longer readily available in the US. Recent consensus recommendations suggest that apomorphine therapy can be initiated using a slower titration scheme without antiemetic pre-treatment. 26 In the outpatient setting, we initiated CSAI with a 2 mg bolus dose and initial 1 mg/h infusion and utilized a gradual titration schema (0.5–1.0 mg/h increments). This may have allowed tolerance to nausea to develop. Only 3 patients discontinued due to nausea, and patient-reports of nausea were mild or moderate in severity.
Unlike dopamine receptor agonists that act predominantly on the D2-family of receptors (D2, D3 and D4), apomorphine has dual D1-family (D1 and D5) and D2-family receptor affinity that is similar to dopamine. 18 Apomorphine also does not display selectivity for D3 relative to D2 receptors.18,27 It has been suggested that D3 (relative to D2) selectivity may be responsible for the increased risk for ICDs encountered with pramipexole, ropinirole and rotigotine. 27 Apomorphine has been reported to have a lower tendency to cause ICDs.27,28 Of the 11 patients who had reported ICDs in this study, 9 were concomitantly receiving one of these D3-selective dopamine receptor agonists.
This study was designed to illustrate the benefits of using adjunctive CSAI in US patients with PD in an outpatient setting. Strengths of this study include its “real world” clinically enrolled patient population, prospective 52-week study duration, and use of CSAI as an adjunctive (as opposed to replacement) therapy that was added onto optimized levodopa therapy with at least one other adjunctive medication. Important limitations of this study include its open-label design with lack of a control treatment arm. In addition, there was possibility of selection bias, including enrichment in enrollment for patients with more severe motor fluctuations. We also excluded patients with clinically significant ongoing psychosis or impulse control disorders.
Many individuals with PD have recurrent OFF fluctuations that limit reliable and consistent Good ON time despite optimization of existing medications. The ease-of-use, demonstrated tolerability, and durable efficacy of CSAI support its use at an earlier disease stage than surgical therapies. Results from this US, open-label, prospective, extended study of CSAI reinforce global reports of apomorphine infusion utility as an effective, intermediate treatment option for patients with disabling motor fluctuations despite optimized oral medications.
Supplemental Material
sj-docx-1-pkn-10.1177_1877718X241310727 - Supplemental material for Continuous, subcutaneous apomorphine infusion for Parkinson disease motor fluctuations: Results from the phase 3, long-term, open-label United States InfusON study
Supplemental material, sj-docx-1-pkn-10.1177_1877718X241310727 for Continuous, subcutaneous apomorphine infusion for Parkinson disease motor fluctuations: Results from the phase 3, long-term, open-label United States InfusON study by Stuart H Isaacson, Alberto J Espay, Rajesh Pahwa, Pinky Agarwal, Holly A Shill, Jennifer Hui, Khashayar Dashtipour, Mark Lew, Peibing Qin, Andrea E Formella, Gianpiera Ceresoli-Borroni, Peter A LeWitt and in Journal of Parkinson's Disease
Footnotes
Acknowledgments
The study was funded and conducted by US WorldMeds, LLC, and Supernus Pharmaceuticals, Inc., who participated in the design and conduct of the study, data collection, and data management. We thank the patients and site staff involved in the study, Zara Melyan PhD, (formerly with Supernus Pharmaceuticals, Inc.) and ACP Clinical Communications Ltd (funded by Supernus Pharmaceuticals Inc), for editorial support. We would also like to acknowledge Oyinkansola Odebo, MD, (formerly with Supernus Pharmaceuticals, Inc.) and Zulane Maldonado-Cruz, MD, (Supernus Pharmaceuticals, Inc.) for assistance with safety results reporting; Milan Joshi, Wentao Wu, and Ya Wang, PhD, MS, (Supernus Pharmaceuticals Inc.) for assistance with data analysis; and Thomas Clinch (US WorldMeds) for statistical support during the study and participation in early drafts of the manuscript.
ORCID iDs
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: US WorldMeds, LLC, and Supernus Pharmaceuticals Inc.
Declaration of conflicting interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Stuart H. Isaacson, Peter LeWitt, Alberto J. Espay, Rajesh Pahwa, Pinky Agarwal, Holly A. Shill, Jennifer Hui, and Khashayar Dashtipour were investigators in the InfusON study and report fees for consultancy from Supernus Pharmaceuticals Inc and US WorldMeds, LLC. Gianpiera Ceresoli-Borroni, Andrea Formella, and Peibing Qin, are employed by Supernus Pharmaceuticals Inc. In addition, Dr Isaacson reports honoraria for CME, consultant, research grants, and/or promotional speaker on behalf of: Abbvie, Acadia, Acorda, Addex, Affiris, Alexza, Allevion, Amneal, Annovis, Aptinyx, Athira, Bial, Biogen, BlueRock, Britannia, Bukwang, Cala, Cerecor, Cerevel, CND, Eli Lilly, Enterin, Esteve, Fasikl, GE Healthcare, Global Kinetics, Inhibikase, Intra-Cellular Therapies, Ipsen, Jazz, Kyowa Kirin, Lundbeck, Medscape, Merz, Michael J. Fox Foundation, Mitsubishi Tanabe, Neuralys, Neurocrine, NeuroDerm, Neurolive, Exeltis, Novartis, ONO Pharmaceutical, Parkinson Study Group, Pharma2B, Praxis, Revance, Roche, Sage, Sanofi, Scion, Stoparkinson, Sunovion, Sun Pharma, Supernus, Teva, Theravance, Transposon, and UCB. Dr Espay has received grant support from the NIH and the Michael J Fox Foundation; personal compensation as a consultant/scientific advisory board member for NeuroDerm, Amneal, Acadia, Avion Pharmaceuticals, Acorda, Kyowa Kirin, Supernus (formerly, US WorldMeds), NeuroDiagnostics, Inc (SYNAPS Dx), and Herantis Pharma; and publishing royalties from Lippincott Williams & Wilkins, Cambridge University Press, and Springer. He cofounded REGAIN Therapeutics and is co-inventor of the patent “Compositions and methods for treatment and/or prophylaxis of proteinopathies.” He serves on the editorial boards of the Journal of Parkinson's Disease, Journal of Alzheimer's Disease, European Journal of Neurology, Movement Disorders Clinical Practice, and JAMA Neurology. Dr Pahwa serves as a consultant for Abbott, AbbVie, ACADIA, Acorda, Allevion, Amneal, Appello, Biogen, BioVie, CalaHealth, Convatec, Fasikl, Genetech, Insightec, Jazz, Keiferx, Kyowa, Lundbeck, Merz, Mitsubishi Tanabe, Neurocrine, Ono, PhotoPharmics, PSG, Reagenxbio, Sage, Sun Biopharma, Supernus, UCB. He receives research support from Abbott, AbbVie, Alexza, Amylyx, Annovis, ASK Bio, Attune, Biogen, Bluerock, Bukwang, Cerevance, Cerevel, EIP, Fasikl, GemVax, Global Kinetics, Inhibikase, Intra-cellular Therapies (ITI), Jazz, Michael J Fox Foundation, Neurocrine, NeuroDerm, ONO, Parkinson's Foundation, Praxis, Roche, Sage, Scion, Sun Pharma, Teva, Theravance, UCB. Dr Agarwal has received compensation to serve as consultant and speaker for Acadia, Acorda, Adamas, Amneal, US WorldMeds and reports research grants from the Michael J Fox Foundation, Merck, Astellas, Parkinson Study Group, CHDI Foundation, US WorldMeds, Lilly USA, Biogen Idec, Acadia Pharmaceuticals, Genentech (Roche), Prilenia Therapeutics, Sun Pharma Advanced Research Company Limited (SPARC) and Centogene AG. Dr Shill reports consultancy for Biogen, Acadia, Mitsubishi Tanabe Pharma America, Inc; honoraria for advisory boards from Kyowa Kirin Inc, Jazz Pharmaceuticals and research funding from Barrow Neurological Foundation, Impax Laboratories LLC, US World Meds, NIH, Parkinson Study Group/Biogen, Sunovion, Intec Pharma, Cala Health. Dr Hui reports consultancy for Sunovion. She has research grants from US WorldMeds, Sunovion, and Roche. Dr Dashtipour has received compensation to serve as an advisor and speaker from Allergan, Acadia, Abbvie, Acorda, Amneal, Ipsen, Lundbeck, Neurocrine, Teva and US WorldMeds.
Dr Lew reports consultancy for Acorda, Supernus, Neurocrine, Kyowa, Amneal, UCB, and Global Kinetics. He has research grants from the Parkinson's Study Group, Michael J. Fox Foundation, Anenberg foundation, UCB, Jazz Pharmaceuticals, Neuraly, NIAA, Jazz Pharmaceuticals, Inhibikase Therapeutics, Biogen, Ono Pharma. Dr LeWitt has received compensation from consulting or lectures from Abide, Amneal, Biogen, Bukwang, Cavion, Denali, Hoffmann-La Roche, Jazz Pharmaceuticals, Kyowa, Mitsubishi NeuroDerm Ltd, Neurocrine, ONO Pharma USA, Revance, US WorldMeds, and Voyager Therapeutics; research grant support from Acorda Therapeutics, Biotie Therapies, Lundbeck, Merz, Michael J Fox Foundation for Parkinson's Research, Parkinson Study Group, Pharma 2B, Revance, Roche, Sunovion, Sun Pharma, and Supernus. Peter LeWitt and Alberto Espay are Editorial Board Members of this journal but were not involved in the peer-review process of this article nor had access to any information regarding its peer-review.
Data availability
The datasets generated during the current study are available from Supernus Pharmaceuticals on reasonable request. Supernus Pharmaceuticals, Inc. will share study documents with qualified researchers who provide valid research questions.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.