
Abstract
Technological innovation related to the advent and development of the Next-Generation Sequencing (NGS) has provided significant advances in the diagnosis of disorders with genetic and phenotypic variability, such as neurodegenerative diseases. However, the interpretation of NGS data often remains challenging, although advanced prediction tools have contributed to primarily assess the impact of some missense variants. Here, we report a patient with Parkinson’s disease (PD) and a family history of disease, in which a panel of 29 disease-causing or risk genes for PD were analyzed. We identified a new missense variant in the SNCA gene. Although this variant might be associated with PD in this family, it has been currently classified as a “Variant of Unknown Significance” because of the lack of segregation with disease. Indeed, we subsequently found the same mutation in an unaffected sister. Nevertheless, this finding may help clinicians and researchers in questioning the causative role of genetic variants within the daily clinical and diagnostic settings.
Parkinson’s disease (PD) is a neurodegenerative disorder characterized by the loss of dopaminergic neurons of the substantia nigra, with cytoplasmic α-synuclein inclusions (Lewy bodies). PD typically manifests as asymmetric resting tremor, bradykinesia, rigidity, and postural instability. Autonomic symptoms, behavioural and cognitive changes leading to dementia, and sleep disorders are key non-motor features [1]. To date, at least 23 chromosomal regions and 19 disease-causing genes for parkinsonism, with either autosomal dominant or autosomal recessive transmission, have been identified [2]. Most cases, however, result from an interaction between different genetic and environmental factors [3]. Alpha-synuclein mutations (SNCA gene) are a very rare cause of PD: fewer than 10 missense/nonsense/duplication mutations have been identified and associated with the gene as a cause of autosomal dominant PD; conversely, LRRK2 gene mutations have been more frequently associated with PD. We describe here the identification of a novel variant in the SNCA gene, by using an ad hoc Next-Generation Sequencing (NGS) panel.
A 67-year-old man, presenting with gait impairment started eight years earlier, came to our observation because of a progressive deterioration of his cognitive status, associated to psycho-motor inertia, apatia, abulia, emotional lability, agitation, and visual hallucinations. Hyposmia and REM sleep behavior disorder preceded the onset of symptoms for at least 20 years. Neurological examination revealed hypomimic face, slowing of downwards eye movements, hypophonic voice, small gait disorder, marked axial bradykinesia and rigidity in the right side, resting tremor in the right arm, bilateral palm-chin sign, depressed mood, and anxiety. Overall, the motor symptoms severely affected the patient’s mobility, although they partially responded to the dopaminergic treatment (L-dopa 100 mg+carbidopa 25 mg+entacapone 200 mg, quarter in die; safinamide 100 mg die). A major neurocognitive disorder with mild functional impairment in the activities of daily living was evident at the psycho-cognitive evaluation. Neuroimaging showed mild bilateral atrophy of the parietal and occipital cortices. Both parents and patient’s brother and sister had died. His mother suffered from arterial hypertension and died because of a stroke; his brother and sister had presented with a clinically similar syndrome, being both affected by PD and dementia; no detailed information was available on his father, who was likely not affected by any neurological disorder. Given the positive family history of PD, an 80-year-old sister was subsequently investigated; although she complained memory deficit, no significant cognitive impairment or extrapyramidal syndrome was observed. The study was approved by the Ethical Committee of the Oasi Research Institute – IRCCS, Troina, Italy. A written informed consent was obtained by the subjects involved prior to the beginning of the study.
The genetic panel included the following disease-causing or risk genes for the development of PD: ABCB1, ATP13A2, BDNF, CP, CYP17A1, CYP2D6, DRD2, EDN1, FBXO7, GBA, GCH1, HFE, HGF, HMOX1, IGF1 R, IGF2, IGF2 R, IL6, LRRK2, MAPT, MCCC1, PRKN, PINK1, RAI1, SNCA, SOD1, SOD2, TNF, TAF1. The design target coverage was 98.7%. Amplicon library, emulsion PCR, sequencing runs, data analysis of runs, and Sanger confirmation were performed, as described by Calì; et al. [4]. Data of runs were processed using the Ion Torrent Suite 5.0, Variant Caller 5.0, Coverage Analysis 5.0 (Thermo Fisher Scientific), the Ion Reporter (Thermo Fisher Scientific), and the wANNOVAR tools [5]. Furthermore, the PolyPhen-2, SIFT, and Provean software tools were used to predict the pathogenicity of the mutations, as well as the following function prediction tools into the ANNOVAR: LRT, Mutation Taster, FATHMM, RadialSVM, LR, MutPred, Combined Annotation Dependent Depletion (CADD), and Mutation Assessor. We removed all the common variants (Minor Allele Frequency >1%); reported in the public databases 1000 Genome Project Database and Exome Variant Server. Then, we searched for all the variants listed as pathogenic in the ClinVar, The Human Gene Mutation Database (HGMD Professional 2018.1), ExAC, ESP6500, dbSNP, ClinVar, and GWAS.
Through this NGS “targeted panel” approach, a variant, in heterozygous status, in the SNCA gene (NM_000345.3) c.44T>C (p.Val15Ala) never described before was identified in the proband (Fig. 1). The Valine position 15 of the protein of the SNCA gene is highly conserved during evolution, and the prediction analysis in silico by Polyphen (probably damaging), SIFT (deleterious), PROVEAN (deleterious), and Mutation Taster (disease causing) indicated the possible causative contribution of the mutation to the risk of developing the disease. Furthermore, the ANNOVAR’s list of functional annotations (LRT, MutationTaster, FATHMM, RadialSVM, LR) predicted the variant as deleterious, although the MutPred, CADD, and Mutation Assessor did not (Table 1). Evidence of pathogenicity was also rated as “moderate” within the ACMG Standard Guidelines [6]. Finally, it is worth to mention that this variant was not present in 60 ethnicity-matched healthy subjects. Similarly, the variant was not found in the 1000 Genome Project Database, ExAC, ESP6500, dbSNP, ClinVar, and GWAS. In line with the study on the SNCA variants [7], our patient showed not only PD, but also cognitive decline, depression, psychotic symptoms, anxiety, and disordered sleep.
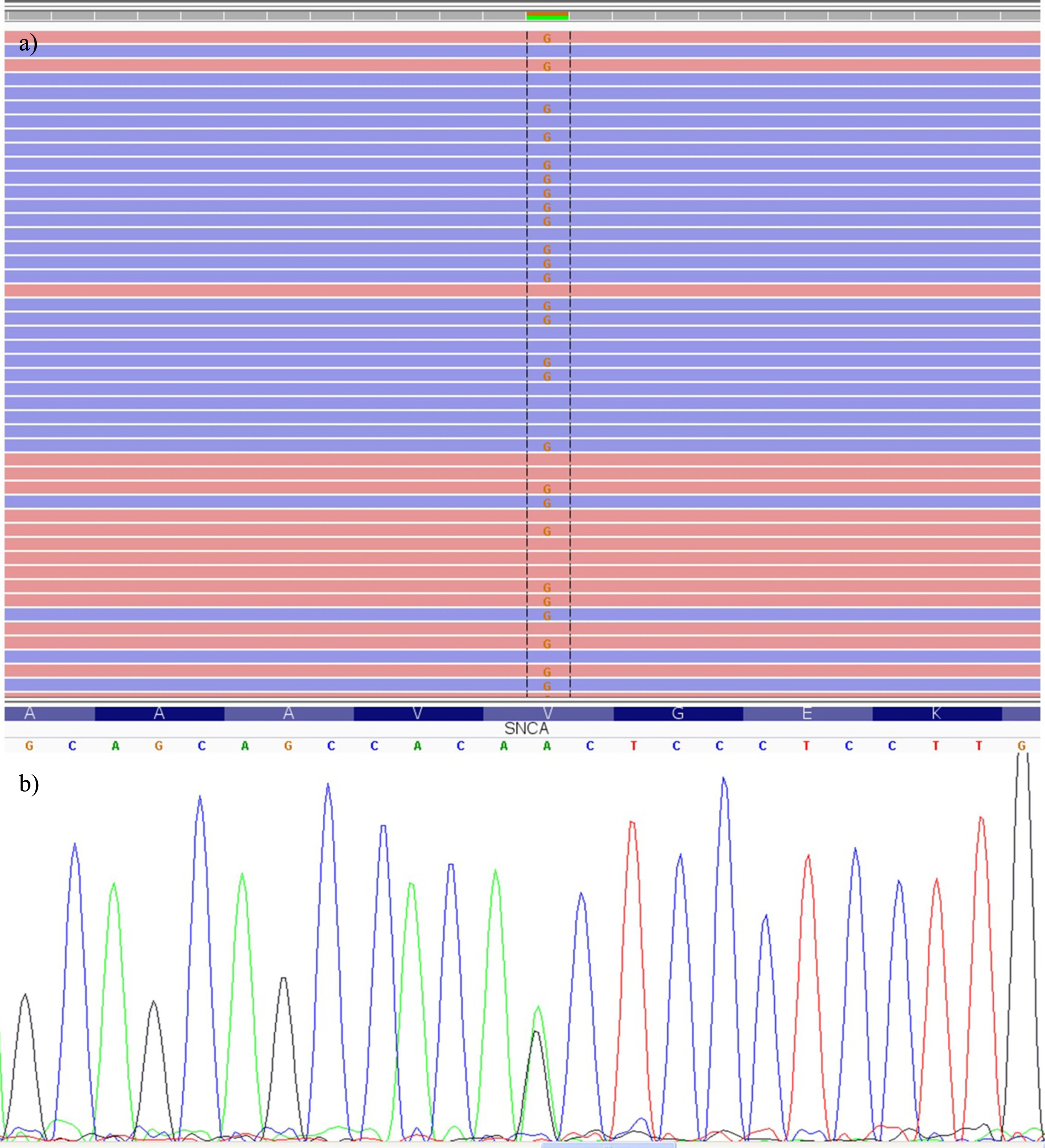
a) Integrative Genomics Viewer visualization. Total read counts 123: A = 60 (49%), G = 63 (51%). b) DNA sequencing electropherogram showing the heterozygous c.44T>C (p.Val15Ala) missense mutation in the exon 2 of the SNCA gene.
In silico functional annotations of the c.44T>C (p.Val15Ala) variation
D, deleterious; L, low.
Although the p.Val15Ala variant may contribute to the pathogenesis of PD in this patient, the pathogenicity still remains uncertain given the pattern of segregation and the possibility of other genetic causes. Indeed, the same c.44T>C (p.Val15Ala) genetic variant was subsequently detected, in heterozygous status, also in his unaffected sister. In addition, a recessive pattern of inheritance was suspected, since both parents were apparently healthy and only siblings were affected. Although the patient did not exhibit any mutation or genetic variants in the other most representative disease-causing or risk genes for the development of PD, other types of mutation (i.e. exon rearrangements, large insertion/deletion) and genes causing recessive PD were not excluded and, therefore, they might still play a role in the phenotypic expression of this patient. For these reasons, this finding has been currently classified as a “Variant of Unknown Significance”. Further studies are necessary aiming at the identification of new mutations in PD genes other than those described in the exons or in the conventional splicing sites transcripts. In this frame, NGS is a major drive in providing a powerful way to study the genetic basis of human diseases, although this technology has still some limitations, mainly related to the large insertion/deletion detection. Moreover, differences of penetrance and expressivity, largely due to modifier genes, environmental factors, allelic variations, complex genetic and environmental interactions, may explain the phenotypic differences observed in this family, and, at the same time, are a challenging question for both clinicians and researchers. Finally, although the in silico analyses are useful to predict the effects that each variant may produce on the transcript, their results should be handled cautiously, and further evidences within the clinical and diagnostic settings are needed before refusing or supporting the pathogenetic role of new variants.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
Ethical compliance statement
The study was approved by the local Institutional Ethic Committee. A written informed consent was obtained prior to the beginning of the study by all the persons involved.
Footnotes
ACKNOWLEDGMENTS
Special acknowledgement for this paper is due to Mrs Eleonora Di Fatta for her valuable assistance in the translation, preparation, and formatting of the text.
This work was partially supported by the Italian Ministry of Health - Ricerca Corrente 2017 - and the “5 per mille” funding.