
Abstract
Parkinson’s disease (PD) is a neurodegenerative disorder that is characterized by loss of dopaminergic neurons in the substantia nigra pars compacta, depletion of dopamine in the striatum and the presence of Lewy bodies. Cancer is uncontrolled growth of cells in the body and migration of these cells from their site of origin to other parts of the body. PD and cancer are two opposite diseases, one arising from cell proliferation and the other from cell degeneration. This fundamental difference is consistent with inverse comorbidity between most cancers and neurodegenerative diseases. However, a positive association of PD and melanoma has been reported which has recently become of significant interest. A link between PD and cancer has been supported by many epidemiological studies, most of which show that PD patients have a lower risk of developing most cancers than the general population. However, the mechanisms underlying this epidemiological observation are not known. In this review we focus on epidemiological studies correlating PD and melanoma and the possible mechanisms underlying the co-occurrence of the two diseases. We explore possible explanations for the important observations that more PD patients develop melanoma that would otherwise be expected and vice-versa.
Keywords
ABBREVIATIONS
BRCA1 Associated Protein 1
v-raf murine sarcoma viral oncogene homolog B1
Cyclin-dependent kinase 4
Cyclin-dependent kinase inhibitor
Cutaneous melanoma
central nervous system
cytochrome p450 debrisoquine hydroxylase locus
dopamine
DCC netrin1 receptor
3, 4-dihydroxyphenylalanine
Epidermal growth factor
Erb-B2 Receptor Tyrosine Kinase
Guanine nucleotide-binding proteinG(q)
glutathione S-transferase M1
Inducible NO synthase
KIT Proto-Oncogene Receptor Tyrosine Kinase
Lewy Bodies
Loss of heterozygosity
Leucine rich repeat kinase
Mitogen-activated protein kinase 1
Melanocortin-1 receptor
Melanogenesis Associated Transcription Factor
Parkinson’s disease
Pten-induced kinase 1
Peripheral nervous system
Phosphatase and Tensin Homolog
Substantia nigra
Substantia nigra pars compacta
Tyrosine hydroxylase
Tumor protein p53
Transient receptor potential melastatin 7
Tyrosinase
Ubiquitin carboxyl-terminal hydrolase L1
Ultraviolet radiation
Vitamin D receptor
INTRODUCTION
Parkinson’s disease (PD) is a neurodegenerative disorder that is characterized by loss of dopaminergic neurons in the substantia nigra pars compacta (SNpc). In addition, PD is associated with depletion of dopamine (DA) in the striatum and the presence of Lewy bodies (LBs) as proteinaceous inclusions in the cytoplasm of surviving neurons. These neurons are melanin positive and the LBs are predominantly composed of fibrillary α-synuclein [1, 2]. Cancer is characterized by two main features: uncontrolled growth of cells in the body and migration of these cells from their site of origin to other parts of the body by a phenomenon known as metastasis. Cancer and PD can be considered as opposite diseases, one arising from cell proliferation and the other from cell degeneration. Despite this apparently fundamental difference, which is consistent with inverse comorbidity between most cancers and neurodegenerative diseases, a positive association of PD and certain cancers has recently become of significant interest [3–6].
A link between PD and cancer has been supported by many epidemiological studies, most of which have shown that PD patients have a lower risk of developing most cancers than the general population. The risk of dying from cancer is lower in PD patients than in the general population. This suggests that, like Alzheimer’s and Huntington’s disease, PD apparently offers some kind of protection against certain cancers. Remarkably, however, PD patients have a significantly higher risk of developing melanoma (HR, 2.75; 95% CI, 1.35–5.59) [7]. The increased risk for melanoma found in this study (3.6; 95% CI, 2.2–5.6) was comparable with what was seen in the DATATOP (3.3; 95% CI, 1.1–7.8) but significantly lower than in the PRECEPT study (20.9; 95% CI, 9.6–39.7) [8]. The mechanisms underlying this epidemiological observation are not known. In this review we focus on epidemiological studies correlating PD and melanoma as well as on the possible mechanisms underlying the co-occurrence of the two diseases. We explore possible explanations for the important observations that more PD patients develop melanoma that would otherwise be expected and vice-versa.
EPIDEMIOLOGY OF PD AND MELANOMA
In the United States, 1.5 million Americans are currently living with PD and 60,000 new cases of PD are diagnosed each year [9]. The risk of developing PD increases steeply over the age of 60 and is around 0.3% at 75–80 years of age [10]. It is the second most common neurodegenerative disease after Alzheimer’s disease. It is expected that due to the general aging of the population, the number of PD patients will double by 2030 [11]. Based on a meta-analysis, it has been reported that prevalence of PD rose from 107/100,000 persons between ages 50 and 59 years to 1087/100,000 persons between 70 to 79 years [12]. PD is more common in men than in women. The male-to-female ratio is about 1.5 [13–15].
Melanoma is a malignant tumor of melanocytes, which are melanin producing cells of the skin. The incidence of melanoma has increased during the last 4 decades in many countries [16]. The incidence of melanoma is increasing continuously and its estimated annual percent change (EAPC) is currently 2.9% per year in the United States [17].
Co-existence of PD and melanoma
A large number of epidemiological studies have reported a co-occurrence of PD in melanoma patients and melanoma in PD patients, and the incidence is significantly higher than expected [16–21]. The largest prospective study of melanoma in PD so far involved a total of 2,106 patients with a mean age of 68.6 years, 85 of whom were receiving levodopa. It was found that in these patients age- or sex-adjusted relative risk of any melanoma was more than seven times than expected [21]. Another large scale study demonstrated that the diagnosis of melanoma is associated with a 50% increased risk of the development of PD [18] and patients with PD had a two-fold increase in risk of developing melanoma subsequently [19]. In another study that included ∼160,000 people who did not have PD, it was found that individuals who had a family history of melanoma were twice as likely to develop PD in comparison to individuals who did not have history of melanoma in the family [20]. A recent clinical study has shown that in Phase I, patients with PD had a 3.8-fold increased chance of having melanoma that pre-existed as compared with controls and in Phase II, patients with melanoma had a 4.2-fold increased risk for the development of PD. Interestingly, patients who had melanoma without PD had a 10.5-fold increased relative risk of dying from metastatic melanoma compared with patients who had melanoma along with PD [22]. It has also been recently reported that the risk for the development of melanoma was higher than expected in the NET-PD LS-1 cohort with an overall standardized event ratio of 3.6 (95% CI, 2.2–5.6) and this result was similar to the risk reported in earlier comparable clinical trial cohorts [8].
Common markers between PD and melanoma
Transcranial sonograms of the midbrain of PD patients commonly show an extended echogenic substantia nigra (SN), a pigmented region of the midbrain, consistent with degeneration of dopaminergic nigrostriatal projection neurons. An abnormally extended echogenic SN was also observed in melanoma patients, supporting a potential underlying pathogenic pathway common to both diseases [23]. Furthermore, melanoma patients exhibiting abnormally echogenic SN demonstrated increased prodromal motor and non-motor features typical of PD, such asymmetric motor slowing, although hyposmia and colour vision disturbance were not frequently observed [24]. Higher echogenicity of SN in melanoma correlated with lower serum iron, as also observed for PD. Parkinsonism was most commonly observed in patients with the lentigo-maligna type of melanoma, typically located at head or neck areas exposed to sun.
PIGMENTATION PLAYS AN IMPORTANT ROLE IN PD AND MELANOMA
Melanin is a term used to describe a group of natural pigments found in most organisms. It is generated by the oxidation of the amino acid tyrosine, followed by its polymerization and is produced in cells known as melanocytes. In human beings, melanin determines the colour of skin and hair. Individuals with darker skin produce more melanin than individuals with a lighter skin. The process by which melanin is synthesized in human skin is called melanogenesis. In the brain, the pigment-bearing neurons within the brainstem and in the SN produce a pigment known as neuromelanin. Abnormalities in melanin are associated with skin cancers like melanoma, whereas abnormalities in neuromelanin are associated with neurodegenerative diseases, such as PD, suggesting that melanin and neuromelanin may play a role in vulnerability for both PD and melanoma. The type and amount of melanin that is produced are determined by pigmentation genes [25, 26] such as those that encode tyrosinase (TYR), located on chromosome 11, 11ql4-q21, and tyrosine hydroxylase (TH), also located on chromosome 11 (11p15.5). These and other genes involved in pigmentation may be a cause for genetic heterogeneity of melanoma [25–27]. The precise mechanisms of how pigmentation genes are involved in melanoma and PD remain to be understood but possible inferences can be made from existing studies. Dopaminergic neurons and melanocytes contain melanin and neuromelanin respectively, but the synthesis of both pigment types begins with tyrosine and proceeds via formation of 3, 4-dihydroxyphenylalanine (DOPA). The pigmentation-related genes that play a role in the synthesis of DOPA and the subsequent product dopaquinone play an important role both in DA synthesis in nigral neurons and in the synthesis of melanin in melanocytes, suggesting that neuromelanin and melanin pathways may constitute a fundamental linkage between PD and melanoma.
Role of melanin in melanoma
The higher incidence of melanoma in light skinned people than in dark skinned populations suggests that melanin may play a protective role in melanoma [28, 29]. This is also supported by the observations that high melanin density is associated with reduced development of melanomas and other cancers that are sun-induced [28] whereas low melanin density such as in the upper arms is linked to increased incidence of melanoma [30]. This protective effect of melanin may be explained by the ability of melanin to absorb or scatter the UVR, dissipating the UVR energy and preventing DNA damage, which leads to malignant melanoma and other skin cancers. It has been reported that levodopa, an intermediate in the biosynthesis of the pigment melanin, selectively inhibits the growth of human melanoma cells but not of non-pigmented control cells [31], suggesting that induction of melanin synthesis correlates with inhibition of cell proliferation [31, 32] and also suggesting another mechanism by which high melanin levels may lead to a reduction in the susceptibility of cells to develop into melanoma Fig. 2.
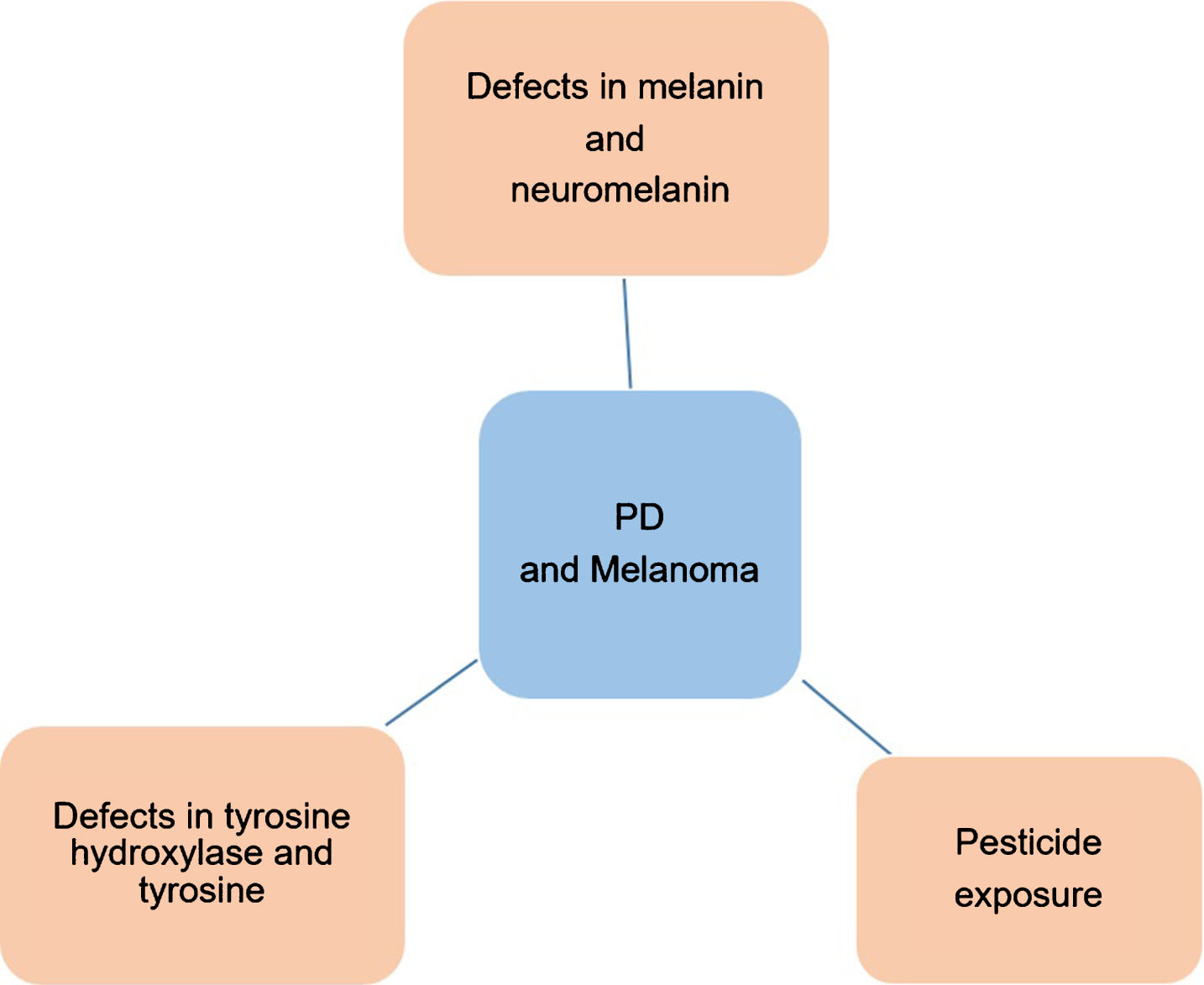
Schematic representation of possible mechanisms that can explain the co-occurrence of PD and melanoma.
Role of neuromelanin in PD
In the SN, neuromelanin is responsible for the dark pigmentation of the dopaminergic neurons. In the brains of PD patients, loss of these dopaminergic neurons causes decreased levels of neuromelanin in the SN, but there has been no correlation reported in the levels of neuromelanin in the SN of PD patients of different race. It has been suggested that neuromelanin formation may be neuroprotective because it can scavenge toxic dopaquinone and help in sequestering metal ions such as copper, iron, cadmium and manganese [33, 34] in human nigral neurons. Lack of neuromelanin makes the neurons more susceptible to oxidative stress [35, 36] and leads to impairment of motor function [35]. The lower occurrence of PD in the black population than in Caucasian populations suggests that, in analogy to the higher cutaneous melanin synthesis, there may be a higher neuromelanin synthesis in the black population, enhancing the protective effects in the SN against external toxic substances [35, 36].
It has also been suggested that melanosis (excessive production of melanin) might protect against both PD and melanoma. This is supported by studies that show a negative association between tobacco smoking and PD [37, 38]. In smokers, melanosis occurs as a result of increased melanin synthesis [39, 40] because nicotine can stimulate activity of melanocytes and production of melanin or the capability of melanin to bind to noxious substances generated by tobacco smoke [39, 40]. Increased melanosis in smoking PD patients therefore may explain the protective effect of smoking against PD.
RISK FACTORS FOR PD AND MELANOMA
Although the exact mechanisms underlying PD are unknown, both genetic and environmental factors can be involved in disease development. Current studies show that mitochondrial dysfunction leading to the production of reactive oxygen species (ROS), impaired clearance of proteins such as α-synuclein from the cytoplasm and induction of inducible NO-synthase (iNOS) in the SNpc are some major mechanisms underlying PD. Mutations in α-synuclein [41], leucine-rich repeat kinase 2 (LRRK2) [42] and possibly ubiquitin carboxy-terminal hydrolase L1 (UCHL1) [43] cause autosomal-dominant forms of familial PD, whereas mutations in parkin [44], DJ-1 [45] and phosphatase and PTEN-induced kinase 1 (PINK1) [46] cause autosomal-recessive forms of hereditary disease. While the above gene products are some of the earliest discovered causes of familial PD, a host of additional genes have been implicated in the etiology of PD in recent years, including many proteins involved in intra-cellular trafficking, auto-phagosomal and lysosomal pathways [48–50].
UV radiation (UVR) exposure is believed to be a major risk factor for melanoma in Caucasians [51] but its role in the darkly pigmented populations like African-Americans, who typically develop melanoma in plantar, palmar, sublingual and mucosal surfaces that are sun protected, is controversial [52]. It is likely that there are factors other than UV exposure that contribute to the development of melanoma [53]. Indeed, several different mechanisms have been linked to the etiology of melanoma in different parts of the body [54], which further suggests that divergent pathways are involved in the process of disease development [7].
Family history of melanoma is associated with an approximately twofold increased risk of melanoma [55]. The genetic factors that increase the risk of melanoma can be divided into high-penetrance genes and low-penetrance genes [56–58]. In families that have a high risk of developing melanoma, mutations in two high-penetrance genes, cyclin-dependent kinase inhibitor 2A (CDKN2A) and cyclin-dependent kinase 4 (CDK4), are the most common cause of susceptibility [56]. Recent studies have shown that development of melanoma is also directly related to changes in low-penetrance genes, such as those coding for melanocortin-1 receptor (MC1R), a protein involved in melanin synthesis; GSTM1 (glutathione S-transferase), an enzyme that catalyzes the detoxification of different compounds including carcinogenic epoxides; and epidermal growth factor (EGF), which is involved in wound healing and mitogenesis [56]. Inactivating polymorphisms at the cytochrome p450 debrisoquine hydroxylase locus (CYP2D6), which codes for another enzyme that may play a role in detoxifying potentially carcinogenic compounds [56], have also been found in melanoma patients at very high frequencies [56] and polymorphisms of the vitamin D receptor (VDR) are also commonly found in melanoma patients [57]. PLA2G6 gene (on 22q13.1) variability has been shown to be associated with melanoma susceptibility [59] and GNAQ, the gene that encodes an alpha subunit of heterotrimeric G proteins, is mutated in 40% of uveal melanomas [60]. The v-raf murine sarcoma viral oncogene homolog B1 (BRAF), encodes a RAS-regulated kinase, B-Raf, that mediates cell growth [61] and is one of the most frequently mutated protein kinases in human cancers, with mutations being particularly common in melanoma.
POSSIBLE MECHANISMS OF THE CO-OCCURRENCE OF PD AND MELANOMA
Epidemiological studies have clearly documented the association between PD but the mechanisms underlying this association remain unknown. Some possibilities are:
Levodopa and melanoma
A number of studies have reported that levodopa may increase the risk of developing melanoma [62, 63] but this still remains controversial. Levodopa is a substrate for tyrosine hydroxylase, a key enzyme in the production of melanin, and melanoma tumor cells are rich in this enzyme. This raises the possibility that levodopa may facilitate tumor growth. There have been several reports demonstrating that melanoma developed in PD patients at a higher-rate rate than expected even before onset of symptoms of PD or the start of dopaminergic therapy. One report of a patient who had PD and developed melanoma after the starting of L-dopa therapy [62] suggested that L-dopa “should not be used in patients with a history of malignant melanoma or undiagnosed skin lesion because it may cause activation of malignant melanoma”. However, systematic analysis of reports came to the conclusion that the co- occurrence of both PD and melanoma is a coincidence rather than causal [64] and that it is unlikely that there is an association between L-dopa and induction or progression of malignant melanoma [65]. The role of levodopa therapy has been debated in the DATATOP clinical trial cohort. Observed cases of malignant melanoma (N = 5) were compared to the published expected values (N = 1.5) for a standard healthy population. The standardized event ratio was 3.3 (95% confidence interval, 1.1–7.8), indicating that incidence of malignant melanoma was higher than expected in this PD cohort. However, there was no association between levodopa treatment and the incidence of melanoma [65]. Collectively, these studies strongly suggest that there is little link between levodopa-therapy and melanoma [63–67].
Common genetic associations link PD and melanoma
Environmental toxins and genes interact and are presumably involved in PD. Lower detoxification enzyme activity may play a crucial role in pathogenesis of PD [68, 69]. The genes CYP2D6 and GSTM1 encode enzymes that have been reported to play important roles in responses of cells to endogenous and/or exogenous reactive intermediates, thereby influencing the susceptibility of an individual to developing PD [70, 71]. The presence of null polymorphism in the GSTM1 gene can result in the lack of expression of the enzyme, and can cause neuronal death that may contribute to PD [71, 72]. CYP2D6 is believed to be involved in PD because of its role in the regulation of drug and toxin metabolism, but studies linking the relation of PD to CYP2D6 are inconsistent [71, 72]. It has also been reported that polymorphisms in VDR are involved in PD pathogenesis [73]. High frequency of polymorphisms of CYP2D6 or the polymorphism of VDR and null for GSTM1 gene, are found in melanoma patients as well, suggesting that variations in CYP2D6 and VDR or GSTM1 genes may increase the risk for both melanoma and PD. In addition it has been shown that mutations in PLA2G6 are also associated with both PD and melanoma [74, 75], suggesting a further mechanistic link between PD and melanoma (Fig. 1A).
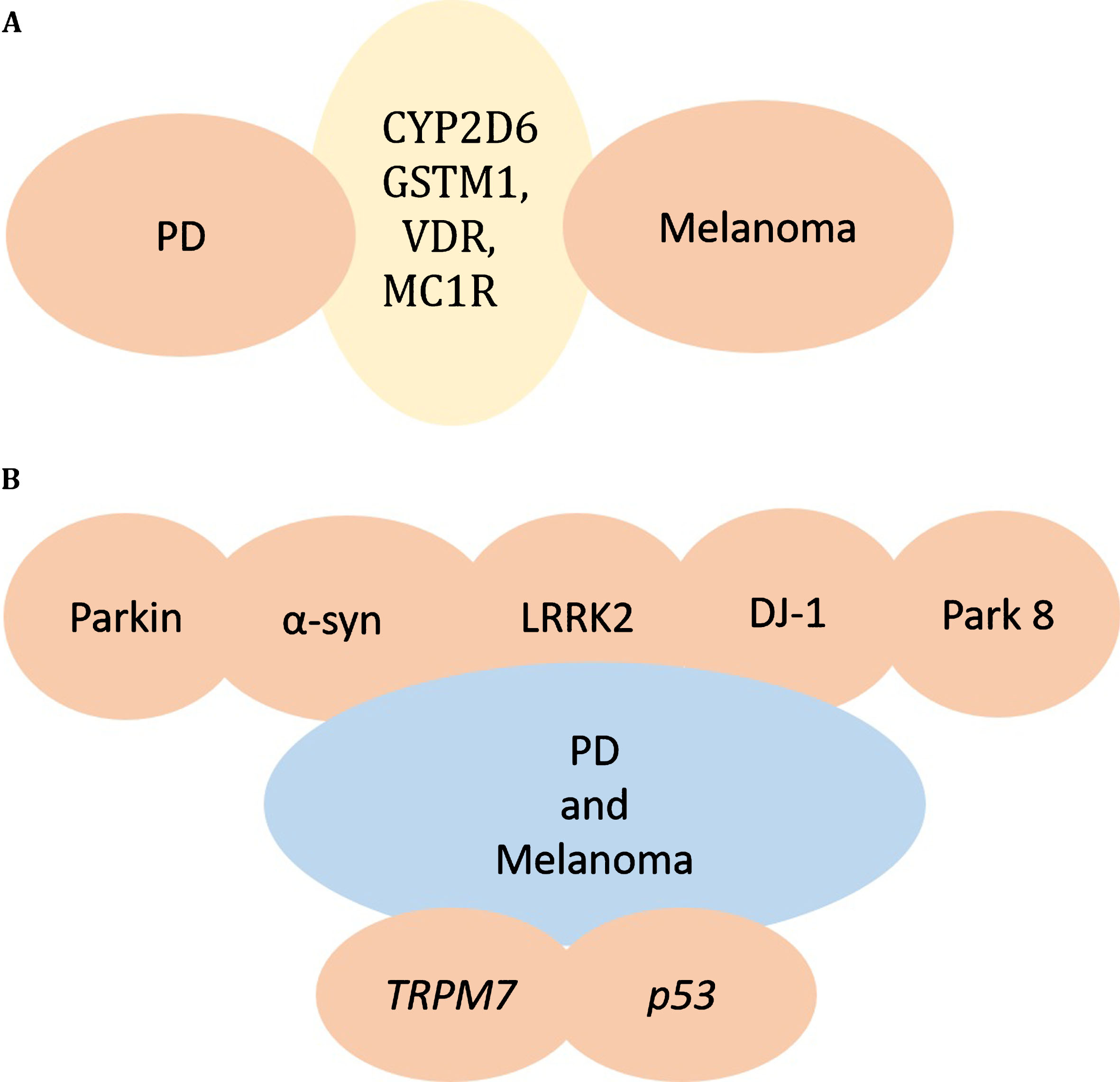
Genes responsible for melanoma and PD. A) CYP2D6 or GSTM1, VDR, MC1R gene alterations are found in both PD and Melanoma, providing a potential link between PD and Melanoma. B) Mutations in parkin, α-synuclein, LRRK2, DJ-1, and other PARK genes may underlie the co-occurrence of PD and Melanoma. TRPM7 and p53 are additional genes altered in PD and melanoma.
Melanoma is strongly associated with a phenotype of fair skin/red hair caused by loss-of-function polymorphisms in the MC1R (melanocortin 1 receptor) gene. One study reported that MC1R is not associated with the co-occurrence of melanoma and PD [76] but another study reported that MC1R variant p.R160W (rs1805008) is marginally associated with PD and modulated the risk of PD in the Spanish population [77]. A recent study has reported that MC1R may be protective in the dopaminergic system, suggesting MC1R as a potential therapeutic target for PD. Since the role of MC1R has already been established in melanoma, MC1R may be involved in a common pathogenic pathway for PD and melanoma [78]. Another possible mechanism linking cutaneous (lentigo-maligna) melanoma and PD may be polymorphisms in the MC3/MC4 melanocortin receptor (MC3R, MC4R) genes which might alter signaling by their ligand, β-melanocyte-stimulating hormone [79, 80].
Links between mutations of PD-related genes and co-occurrence with melanoma
Parkin
It has been reported that mutations in the protein parkin are the most common cause of young-onset PD [81]. Parkin is also considered a candidate tumor suppressor, with its gene located on chromosome 6q25-27 [82]. Many studies have demonstrated that loss of heterozygosity (LOH) in the chromosome 6q25-q27 region is frequently found in many human cancers including melanoma [83, 84], suggesting that parkin mutations could be risk factors for both melanoma and PD. A recent study has analyzed the whole parkin exome/genome from 246 melanoma patients and identified three synonymous mutations, five non-synonymous and one splice region variant in parkin in 3.6% of the samples. Analysis in vitro has showed that wild-type parkin plays a tumor suppressive role in the development of melanoma, causing arrest of the cell cycle, apoptosis and a reduction in metabolic activity. Potential parkin substrates in melanoma were also identified using a mass spectrometry based analysis. It has been shown that the parkin mutation E28K impairs ubiquitination of parkin and abolishes its tumor suppressive activity. Thus data from genomic sequencing together with in vitro data suggest that parkin could be a link between melanoma and PD [44] (Fig. 1B).
Alpha-synuclein
It has been well established that α-synuclein plays a role in PD pathogenesis [1, 85]. α-synuclein accumulates within SN neurons, and some reports indicate that a portion of the protein may be trapped inside granules of pigments during the synthesis of neuromelanin, long before neuromelanin depletion is detectable in PD [86–88]. The peripheral nervous system (PNS) is also affected in PD, as demonstrated by the fact that α-synuclein aggregates are found throughout the nervous system, including the enteric nervous system, sympathetic ganglia, submandibular gland, cardiac and pelvic plexuses, the skin and adrenal medulla [87–89] (Fig. 1B).
The transmembrane protein MART-1 is present in normal melanocytes and is widely expressed in malignant melanoma. MART-1 is the most useful histological biomarker to diagnose melanoma when compared to HMB-45 and S-100 proteins. HMB-45 is not expressed by 20–40% of metastatic melanomas and S-100 is less specific as it is expressed in other malignant tumors [90–92]. It has been shown that α-synuclein shows positive immunoreactivity in both MART-1-positive and negative melanoma cells in many cases, suggesting that α-synuclein might be another good histological biomarker for the diagnosis of malignant melanoma [93].
Several studies have reported that α-synuclein is robustly expressed in melanoma cell lines, primary and metastatic melanoma tissues and nevus tissues [93, 94], suggesting that α-synuclein could play a role in the pathogenesis of melanoma and further suggesting that α-synuclein may cause an increase in the risk for both PD and melanoma.
LRRK2
It has been reported that abnormally high kinase activity of mutant LRRK2 mediates neuronal toxicity and cell death in PD [95]. The B-raf kinase, which is believed to cause a significant proportion of malignant melanoma [61, 96], bears significant similarity to the LRRK2 kinase domain, implying that there could be functional analogies between the activation of the B-raf kinase in melanoma and activation of LRRK2 kinase in PD-associated neurodegeneration [75] Fig. 1B.
DJ-1
DJ-1 was identified as an oncogene and mutations in DJ-1 cause familial PD in an autosomal recessive manner, implying loss of function. It has been shown that DJ-1 is overexpressed in cancers such as melanoma and breast cancer [69] raising the possibility that that DJ-1 mutations associated with PD may act as a protective factor for developing melanoma in PD patients (Fig. 1B).
Exposure to pesticides
There is a well-known link between pesticides that target the mitochondrial electron transfer pathway and PD, including rotenone and paraquat [97]. It has been shown that paraquat and rotenone increase endogenous α-synuclein in SH-SY5Y cells as well as in SK-MEL-2 cell lines and also increase α-synuclein levels in virally transduced SH-SY5Y and SK-MEL2 cells overexpressing WT or A53T mutant α-synuclein, suggesting that exposure to pesticides can play a role in development of PD and melanoma simultaneously [98] (Fig. 2).
The role of rare variants in PD and melanoma
Rare variants analysis across all genes, per gene, or for any individual variant does not show any significant trends among different carrier frequencies between controls and PD, in different models of inheritance, in the following cutaneous malignant melanoma (CMM) risk genes: DCC, BAP1, KIT, ERBB4, MAPK2, PTEN, TP53 and MITF. TYR p.V275F which is a rare variant, and is a pathogenic allele for recessive albinism, was found to be more common in PD cases than in controls, suggesting that this might be a new gene in the dopamine-biosynthetic pathway that could play a role in susceptibility to PD and, since pigmentation genes contribute to CMM, possibly contribute to a link between PD and melanoma [99].
Transient receptor potential melastatin 7 gene
Transient receptor potential melastatin 7 (TRPM7) is a cation channel that is nonspecific and has an enzyme domain [100]. It is expressed in melanoma cell lines, and is involved in melanocyte survival by detoxification of intermediates formed as a result of melanogenesis [101, 102]. A heterozygous variant of TRPM7 has been reported in some PD patients with dementia and some Guamanian patients [103] (Fig. 1B).
Tumor suppressor gene p53
The tumor suppressor gene p53 has been shown to play a role in human neurodegenerative disorders like PD. Therefore, chemical inhibitors of p53 may be effective in suppressing the neurodegenerative process in PD [104]. Loss of p53 function occurs in many human tumors including melanoma [105]. Therefore, it is possible that there may be reduced risk for PD in melanoma patients who harbor the inactivating p53 mutations. Factors like p53 that promote one disorder while protecting against the other may explain the fact that comorbidity between PD and melanoma is imperfect (Fig. 1B).
ENZYMES: A LINK BETWEEN PD AND MELANOMA
Role of tyrosine hydroxylase in PD and melanoma
The enzyme tyrosine hydroxylase (TH) is involved in the conversion of tyrosine to DOPA, the rate-limiting step in the biosynthesis of DA within dopaminergic neurons [106, 107]. In PD, there is a loss of TH in the striatum. The most effective treatment of PD is with the use of DOPA, DA agonists, or inhibitors of DA catabolism. However, an excess of cytosolic DA can form reactive intermediates, such as dopaquinone and reactive oxygen species that are associated with dopaminergic neuronal cell loss. Thus TH, an essential enzyme critical for synthesis of DA, also facilitates the formation of ROS that can cause oxidative stress leading to neuronal loss in PD [106]. TH is also found in human melanoma cells [108]. In skin melanocytes, TH is involved in catalyzing the conversion of tyrosine to DOPA, thereby generating the substrate and activator for tyrosinase, the enzyme that catalyzes the first and rate-limiting step in the biosynthetic pathway of melanin [108] Fig 2.
Role of tyrosinase in PD and melanoma
Tyrosinase (TYR) is a copper-dependent rate-limiting enzyme in melanin synthesis within the melanosomes in the skin [109]. It has been shown that the activity of TYR is low in white skin and high in black skin, and it also controls melanogenesis [109, 110]. In the biosynthetic pathway of melanin, tyrosinase is involved in catalyzing several reactions, including the hydroxylation of tyrosine to DOPA [111]. Tyrosinase is also involved in the oxidation of DOPA to dopaquinone, which then leads to the formation of melanin, eumelanin and pheomelanin [110, 112]. A number of reactive intermediates, such as dopaquinone, which are generated during the process of pigmentation, and formation of melanosomes which are catalyzed by TYR, are involved in the induction of melanocytotoxicity [110, 112]. Although melanocytotoxicity may lead to the inhibition of growth in melanoma cells, the exact role of TYR in the process of malignant change is not well understood since the activity of TYR varies among different melanoma tissues [113]. In most pigmented melanomas, positive TYR activity has been reported, whereas negative TYR activity occurs in both pigmented and non-pigmented melanomas [113]. Thus the enzymatic activity of TYR does not appear to reflect the behavior of the tumor cells, suggesting that TYR activity may not robustly predict the degree or malignancy of melanoma. It has been shown that TYR is also involved in the synthesis of neuromelanin in SN of mice and humans [113, 114]. In the mouse brain, the TYR promoter is active throughout the brain during development, particularly in the SN of adult brain [115]. A recent study has also shown that secretion of neuronal TYR is also involved in the inhibition of apoptosis [116] Therefore the overexpression of TYR in brain can play either a neuroprotective role because of the formation of neuromelanin [33] or play a toxic role due to the formation of melanin precursors, such as DOPA and dopaquinone [117] Fig. 2.
α -synuclein: Role in pigmentation
α-synuclein is highly expressed in melanoma cells, but those cells that express high levels of α-synuclein generate very low levels of or no melanin pigment [114], which can be explained by α-synuclein’s role in the reduction of tyrosine hydroxylase activity [107, 118]. Therefore it can be hypothesized that in the melanocytes from the skin of patients suffering from PD, increased α-synuclein causes inhibition of TH, which leads to decrease in melanin synthesis [109] leading in turn to the increasing the risk for melanoma.
DA neurons in SN and melanocytes in skin both express tyrosinase. When excess α-synuclein accumulates under pathological conditions in DA neurons, tyrosinase may interact directly with α-synuclein, causing local modifications that hinder the physiological functions of α-synuclein [119] and leading to toxic processes, such as increases in DA and dopaquinone levels in the cytosol, which then lead to injury of mitochondria [120] and eventual death of neurons [121]. Furthermore, increased levels of α-synuclein can interact with dopaquinone or other DA metabolites to form intermediates that are toxic, leading directly to neuronal cell death [121, 122].
AUTOPHAGY DEFICITS IN PD AND MELANOMA
Autophagy is the process by which cells maintain cellular homeostasis by digesting damaged proteins or cellular organelles that are no longer required; the word means “self-eating”. Defects in autophagy have been linked to neurodegenerative diseases such as PD [47–49] and increasing autophagy has been suggested as a novel therapeutic strategy for PD that could increase the clearance of aggregated proteins and organelles such as damaged mitochondria [48, 123]. Recent studies have suggested that autophagy may be important for tumors [50, 123–125] by demonstrating that increased autophagy plays a suppressive role in tumor initiation, but promotes the survival of tumors that are already established. Such established tumors seem to use increased autophagy in order to survive periods when the tumors undergo hypoxic or metabolic stress [124]. A recent report suggests that melanoma cells exhibit high levels of autophagy [126]. In contrast, however, defective autophagy has also been reported in melanoma as demonstrated by decreased expression of autophagy genes such as LC3 and Beclin1 [125]. Decrease of autophagy may also lead to impairment of antigen presentation, leading to melanoma immune escape and thereby promoting progression [127]. Interestingly, a compound targeting alpha-synuclein has recently been shown to kill melanoma cells by dysregulating autophagy [127]. The precise roles of autophagy in both PD and melanoma remain to be more thoroughly explored, but autophagy deficits may provide mechanistic clues into the correlation between the two diseases.
CONCLUSION
Epidemiological studies clearly demonstrate that melanoma occurs more frequently among patients with PD and vice versa, and the increased risk of melanoma in PD patients does not appear to be dependent on dopaminergic therapy. Mutations or other alterations in a number of genes/proteins are common to both PD and melanoma, providing potential mechanistic links between the two diseases. These include factors that contribute to cellular detoxification, to melanin biosynthesis, to oxidative stress response, and to cellular trafficking pathways. In addition, defects in cellular processes such as autophagy and protein homeostasis may also be involved in disease co-occurrence. Changes in melanin pigmentation and/or the enzymes that are involved in synthesis of melanin are a key area where these otherwise disparate diseases intersect, and further exploration of the significance of these pathways in both diseases is warranted. Nevertheless, both melanoma and PD are clearly multifaceted disorders with both genetic and environmental risk factors contributing to their development, and other genetic and biochemical factors that act to reduce risk. It will be important to understand the various mechanisms by which these risk factors can be responsible for the increased risk of melanoma in PD patients and vice versa, as well as the fact that this co-morbidity is not inevitable. To this end, it will be useful to develop assays in vitro and in vivo to clarify the consequences of altered pigmentation, specific genetic changes, abnormal autophagy and other pathways that may underlie the co-existence of the these two critically important diseases and drive their incidence and progression.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Footnotes
ACKNOWLEDGMENTS
This work was supported by the Appel Alzheimer’s Disease Research Institute, NIH grants R37AG019391 and R01GM117518 (D.E.).