
Abstract
Background:
Mutations in the leucine rich repeat kinase 2 (LRRK2) gene are among the most common genetic causes of Lewy body Parkinson’s disease (PD). However, LRRK2 mutations can also lead to a variety of pathological phenotypes other than typical PD, including relatively pure nigrostriatal cell loss without alpha-synuclein-positive Lewy bodies or Lewy neurites, progressive supranuclear palsy (PSP), and multiple system atrophy (MSA). The mechanisms behind this remarkable pleomorphic pathology are currently unclear.
Objective:
To genetically and pathologically characterize a case with a LRRK2, p.Ile1371Val rare variant and pathologically proven MSA.
Methods:
From the brain donation program at the Parkinson’s Institute and Clinical Center, we selected 26 brains with family history and a with clinicopathological diagnosis of PD (n = 20), MSA (n = 4), or PSP (n = 2). We performed neuropathological evaluation, including alpha-synuclein and tau immunohistochemistry and sequenced 188 genes that have been reported as causative for or associated with neurodegenerative diseases.
Results:
We identified a known LRRK2, p.Ile1371Val genetic variant in a case with clinically diagnosed and pathologically proven MSA. Neuropathology revealed that the olivopontocerebellar system was more affected than the striatonigral system.
Conclusions:
Our data suggest that genetic variants in the LRRK2 gene can present clinically and neuropathologically as MSA. One other LRRK2 genetic variant (LRRK2, p.Ile2020Thr) has been reported with a neuropathological diagnosis of MSA. Interestingly, the LRRK2 variant (LRRK2, p.Ile1371Val) identified here has been reported previously in a postmortem case with Lewy body PD.
Future studies are critical to discover the mechanisms leading to different neurodegenerative trajectories both in neuronal and glial cell populations.
Keywords
INTRODUCTION
Rare pathogenic and common variants in the LRRK2 gene are known to cause or are predicted to increase risk for Parkinson’s disease (PD) [1]. Several founder mutations have been reported (p.Arg1441Cys, p.Arg1441Gly, p.Gly2019Ser), accounting for about 1–2% of apparently sporadic and 4–6% of familial cases. Some variants are prevalent only in individuals of Asian ancestry such as p.Arg1628Pro and p.Gly2385Arg [2], and a large study investigating 121 LRRK2 variants in 15,000 clinically diagnosed PD cases and controls established causation for additional missense variants in different ethnic populations [3].
Neuropathological case descriptions have been published for only seven LRRK2 mutations, p.Ile1371Val, p.N1437H, p.R1441C, p.R1441G, p.Y1699C, p.Gly2019Ser, and p.Ile2020Thr (reviewed in [4]). Interestingly, LRRK2 parkinsonism does not present neuropathologically with Lewy body PD in all cases. A variety of pathological phenotypes including nigrostriatal cell loss without alpha-synuclein-positive inclusions or rare cases of progressive supranuclear palsy (PSP), and multiple system atrophy (MSA) [1, 5–7] are described in literature. The mechanisms behind this remarkable pleomorphic pathology however remain elusive.
In a recent retrospective study of 37 published and unpublished LRRK2 autopsy cases with different allelic variants, only 17 cases presented with Lewy body pathology with a clinical history of cognitive impairment/dementia, anxiety, and orthostatic hypotension, whereas 20 cases did not show Lewy body pathology [8].
In this report, we describe a rare case of autopsy-proven MSA with a LRRK2 variant (c.4111 A>G, p.I1371V, rs17466213), in which the olivocerebellar system was predominantly affected. This rare variant has been reported before in a case with neuropathologically typical alpha-synuclein-positive Lewy body pathology [9] and has been described in several other clinical cases with PD [10–13].
MATERIALS AND METHODS
Sample ascertainment and DNA isolation
Donors were enrolled through the brain donation program at the Parkinson’s Institute and Clinical Center. DNA was extracted from fresh frozen brain tissue with Qiagen DNeasy Blood & Tissue Kit or from formalin-fixed paraffin embedded tissue with Gentra Puregene Tissue Kit according to manufacturer’s instructions.
188-gene panel sequencing
A targeted 188-gene panel including genes that have been described to harbor pathogenic mutations for or that were associated with Parkinson’s disease, amyotrophic lateral sclerosis, and Alzheimer’s disease was used, and all exons and exon-intron boundaries (Supplementary Table 1) were sequenced. We used Agilent SureSelectTM® for target gene enrichment, and next-generation sequencing was performed on Illumina’s MiseqTM® instrumentation. The raw data were aligned using GATK workflow for pre-processing, variant discovery, and call-set refinement [14]. After variant processing, sequence variants were annotated using Variant Effect Predictor toolkit [15] and presented in Supplementary Fig. 1 as gene-interaction network for this patient with LRRK2, p.Ile1371Val. The LRRK2, p.Ile1371Val mutation was confirmed by bi-directional Sanger sequencing of amplified PCR products from genomic DNA.
RESULTS AND CASE DESCRIPTION
Next-generation sequencing of familial cases with parkinsonism revealed a LRRK2, p.Ile1371Val variant in a case with pathologically confirmed MSA showing primarily olivopontocerebellar involvement (Fig. 1). No other known pathogenic mutations causative for PD, MSA, or other neurodegenerative diseases were detected. This is only the second reported case with autopsy proven MSA in a patient with a LRRK2 gene variant.
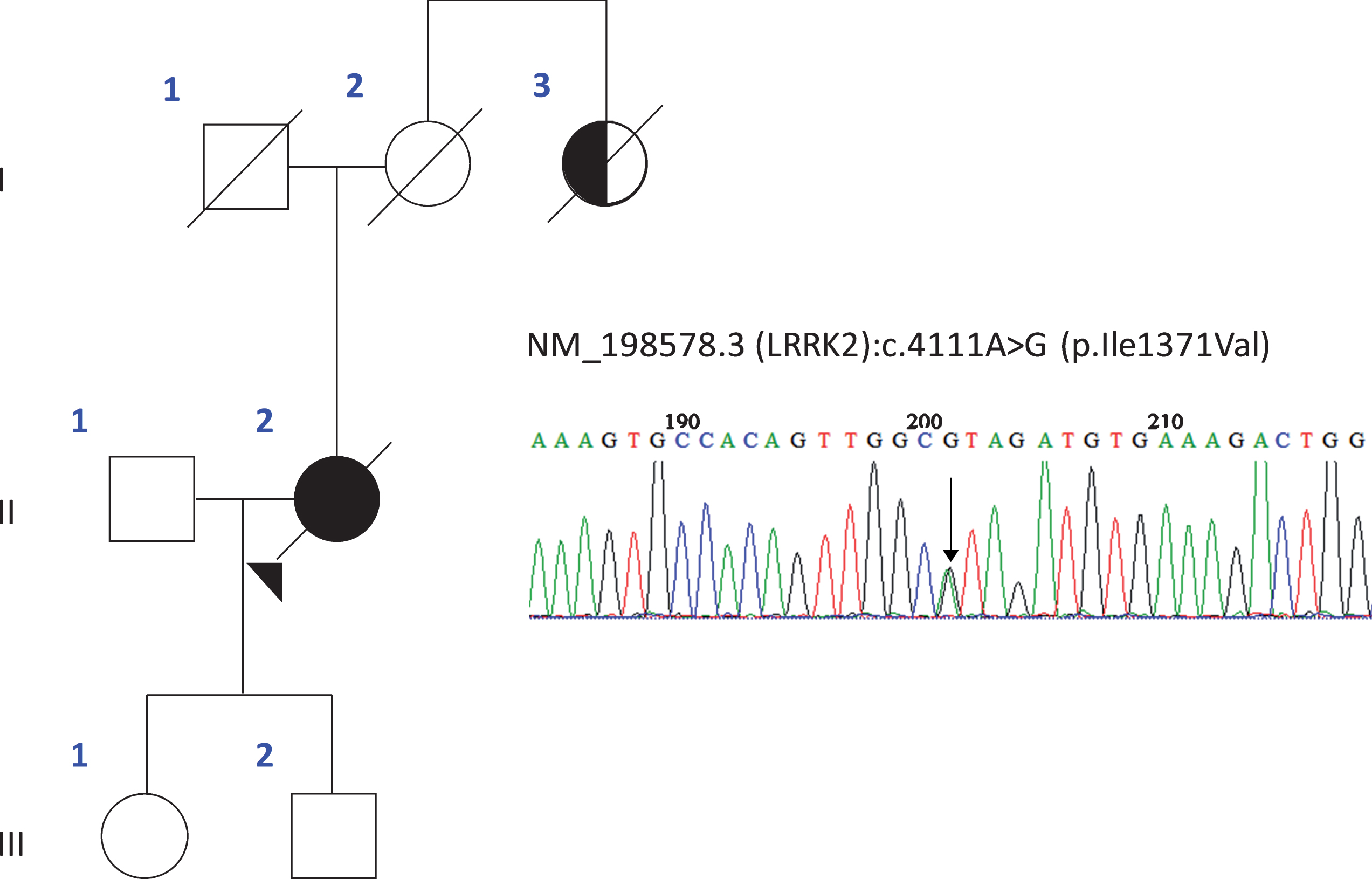
Pedigree and electropherogram. A) Circles in the pedigree indicate female gender, squares indicate male gender, full black symbol indicates MSA, half black symbols indicate PD, but only historical information available. B) Electropherogram from Sanger sequencing. Boxed sequence indicates mutation and change in amino acid sequence from isoleucine to valine.
Patient history and neuropathological presentation
At the age of 50, the patient started experiencing balance problems, slurred speech, and changes in her hand writing. Her disease progressed quickly over a year and a half with severe balance problems resulting in multiple falls, bladder urgency, anxiety attacks, and depression. L-Dopa treatment did not improve her symptoms.
On examination, she arose from a chair slowly, walked with ataxic broad-based gait and without assistance, but with a threat of falling. Finger-to-nose and heel to shin testing were ataxic with mild side to side dysmetria including frequent overshoot ataxia. Rapid alternating movements were slowed bilaterally (left greater than right). No rigidity and no resting, postural, or kinetic tremor noted. Strength was normal, reflexes slightly brisk, but no pathological reflexes or dystonia noted. An MRI was reported to be normal. Symptoms deteriorated over the course of three years and she passed away at age 53. Her maternal aunt was reported to have had parkinsonism.
At autopsy, multiple system atrophy (olivopontocerebellar degeneration) was neuropathologically described. The neocortex showed no neuronal loss, vacuolar changes, or gliosis. The hippocampus, nucleus basalis of Meynert, and hypothalamus had a normal neuronal population, no neuronal cytoplasmic inclusions (NCIs), but exhibited glial cytoplasmic inclusions (GCIs). By alpha-synuclein immunohistochemistry many GCIs, few NCIs and dystrophic neurites were detected throughout the basal ganglia, especially in white matter tracts in the posterior putamen (Fig. 2a, b). There was remarkable neuronal loss in the substantia nigra with extraneuronal neuromelanin, gliosis, numerous GCIs, and few NCIs (Fig. 2c, d). Furthermore, GCIs were frequent in the midbrain gray matter and cerebral peduncle. The inferior olivary nucleus had neuronal loss and gliosis with many NCIs and dystrophic neurites (Fig. 2 g, h). No Lewy bodies were detected, but neuronal loss and extraneuronal neuromelanin were observed. The pontine nuclei had numerous NCIs and neuronal intranuclear inclusions (NIIs) and alpha-synuclein positive dystrophic neurites (Fig. 2e, f). Lastly, there was cerebellar Purkinje cell loss with Bergmann gliosis and Purkinje cell axonal torpedoes, with extensive demyelination in the cerebellar white matter with many GCIs (Fig. 2i, j). No senile plaques or neurofibrillary tangles were detected in the neocortex, hippocampus, amygdala, basal nucleus of Meynert, basal ganglia, or hypothalamus.
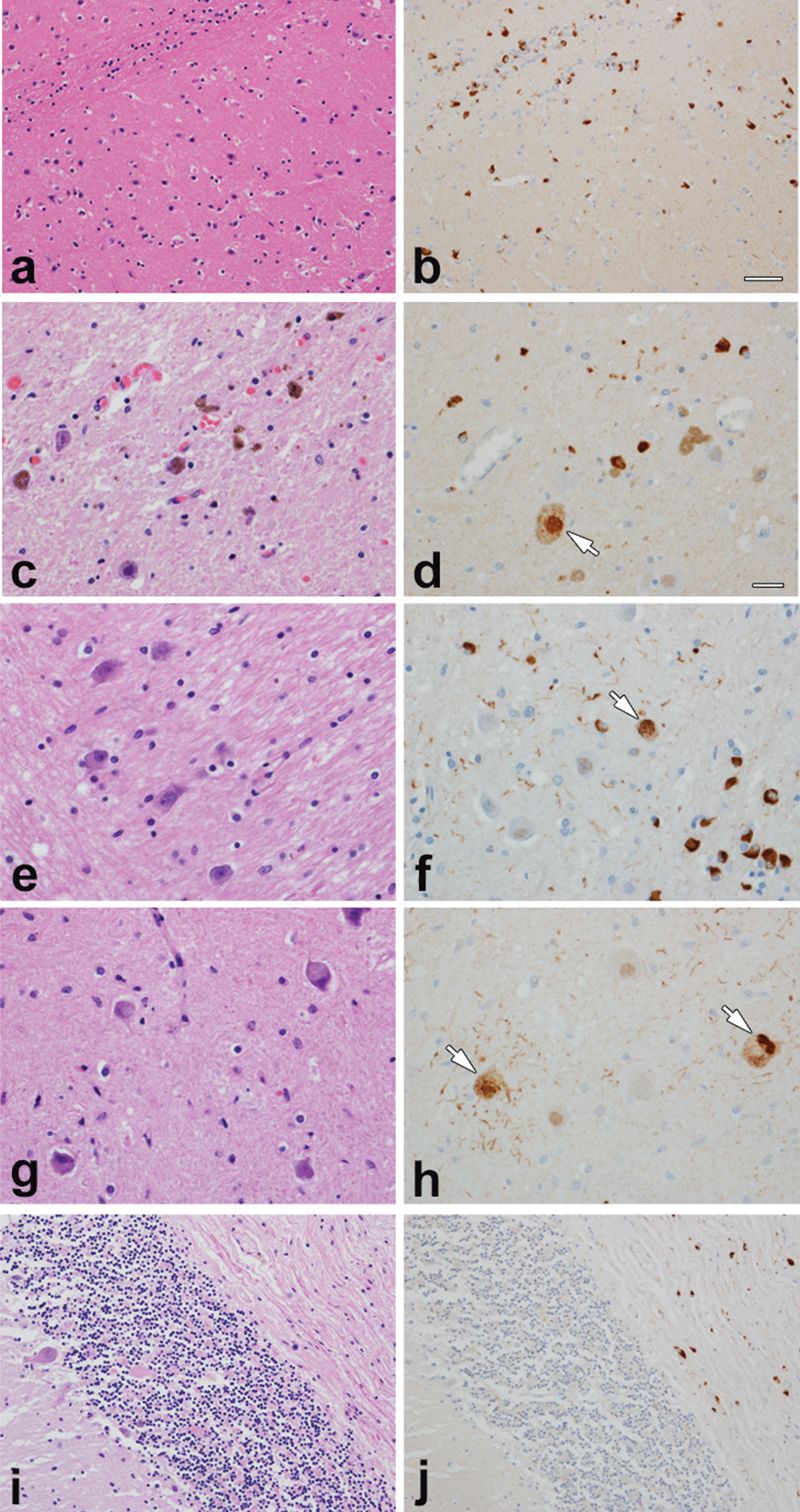
Histopathology of MSA case. Illustrated is pathology typical of MSA affecting both striatonigral and olivopontocerebellar systems, with many glial cytoplasmic inclusions in all affected areas and neuronal cytoplasmic inclusions in select areas. There is neuronal loss in the substantia nigra with extraneuronal neuromelanin. There is marked demyelination in pons and cerebellum. a & b – posterior putamen, c & d – substantia nigra, e & f – pontine base, g & h – inferior olivary nucleus, i & j – cerebellum. Images a, c, e, g, i = H&E stain; b, d, f, h, j =α-synuclein immunohistochemistry. Arrows in d, f, h indicate neuronal cytoplasmic inclusions. Scale bar in b represents 50μm (applies to a, b, i and j) bar in d represents 20μm (applies to c, d, e, f, g, h).
DISCUSSION
Here we report a rare LRRK2 variant (LRRK2, p. Ile1371Val) with a clinical and neuropathological presentation of MSA with primarily olivo-cerebellar involvement and GCIs.
While two large case-control studies in sporadic and familial PD did not find an association with the LRRK2, p. Ile1371Val variant in Caucasian, Asian, or Arab-Berber populations [3, 14], we reviewed ten clinical cases reported in the literature from five families that could indicate pathogenicity of this LRRK2 variant (Table 1).
Clinical information for PD patients with LRRK2, p.Ile1371Val variant
aAS, asymmetry at onset B, bradykinesia; PI, postural instability; R, rigidity; T, tremor. bMMSE, Mini-Mental State Examination; MoCA, Montreal Cognitive Assessment. Note: Seki et al. reports a PD case, but without any clinical description [15].
The LRRK2, p. Ile1371Val variant was first reported in 2005 in a family (case 4862) from East India with 2 affected first degree relatives (parent – child) with typical clinical presentation and good response to dopaminergic therapy. A detailed segregation analysis of additional family members was not possible, but this allele was not detected in 180 controls from North America [10] (Table 1). The LRRK2, p. Ile1371Val variant was next reported in an Italian family (MI-007) and showed segregation in two first degree relatives with asymmetric onset of symptoms and good response to dopaminergic therapy [11]. Autopsy results in one case (MI-007-03) showed typical Lewy body pathology [9]. In that study, one control was found to carry the LRRK2, p. Ile1371Val variant out of a total of 416 control cases. Two additional families with the LRRK2, p.Ile1371Val variants (Family C and Family D) were described with a total of five affected individuals. The clinical presentation as it can be extracted from the clinical description was compatible with typical PD, with the variant segregating in the two families [12]. Lastly, one sporadic PD case from Serbia was described by Jankovic et al. [13] and another case without any detailed clinical description was mentioned in Seki et al. [15]. These descriptions of clinical/pathological phenotype point towards a high-risk variant in familial PD, although co-incidence of this rare variant cannot be excluded.
While MSA presents with glial inclusions as a key pathological hallmark, in the case described here, we also detected frequent alpha-synuclein-positive neuronal inclusions (both cytoplasmic and nuclear) in the substantia nigra, pontine nuclei, inferior olivary nucleus, and locus ceruleus (Fig. 2a-d). In addition, neuritic pathology was detected in the most severely affected areas, especially the putamen, pontine base and inferior olivary nucleus (Fig. 2a-d).
The same genetic variant, LRRK2, p.Ile1371Val, has been previously reported in an autopsy case with a pattern consistent with alpha-synuclein-positive Lewy body pathology affecting primarily the nigrostriatal system [9] (Table 1: patient ID MI-007-03).
Another example for the same allelic variant but different underlying pathologies is presented in the Japanese Sagamihara family with a LRRK2, p.Ile2020Thr mutation. Autopsies from eight affected mutation carriers presented in one case with multiple system atrophy (MSA-P, case 6), one case with typical Lewy-body pathology (case 10), whereas six cases in this family presented with pure nigral degeneration but no Lewy bodies [16].
Functional data show that the kinase activity for the LRRK2, p.Ile1371Val has been slightly increased to a similar extent as the pathogenic mutation LRRK2, p.Arg1441Cys [17].
It is not clear how the same genetic variant produces a different clinical and neuropathological pattern, but as in this case alpha-synuclein accumulation was found both in neuronal and glia cell populations throughout the brainstem [18], this could be indicative of a common underlying disease pathway. Of note, GCIs are also described in sporadic PD [19] as well as in cases with an alpha-synuclein gene triplication [20] while NCIs, as in our case, are described in MSA [21].
In conclusion, we describe the first case with a LRRK2, p.Ile1371Val variant and clinical and neuropathologically proven MSA with both prominent alpha-synuclein-positive glial and neuronal cytoplasmic inclusions. This raises the possibility, that changes in LRRK2 function could be the common underlying cause for an increase and accumulation of alpha-synuclein in neuronal and glia cell populations.
AUTHOR CONTRIBUTIONS
B.S. designed the study and prepared the first draft of the manuscript. J.W.T. clinically evaluated the patient. K-DN, CS, ML performed sequencing and analysis of the 188-gene panel. A.F. extracted DNA from formalin-fixed paraffin embedded tissue blocks for sequencing analysis. FZ performed quality control and curated all DNA samples for the sequencing analysis. D.D. performed, interpreted, and documented brain autopsy and prepared. K.L. reviewed the clinical chart and drafted the case description. K.L. and J. S. performed Sanger sequencing of the LRRK2 mutation. All authors reviewed and edited the manuscript.
CONFLICT OF INTEREST
K-DN, ML, and CS are employees of Biogen Idec. Other authors declare no conflict of interest.
Footnotes
ACKNOWLEDGMENTS
We are indebted to the family for their participation in the Parkinson’s Institute brain donation program. Dr. Lysia Forno (1918–2015) performed the initial neuropathology evaluation of the brain. We thank our summer interns Gautam Bordia, Raghav Bordia, and Bastian Schüle for their extensive clinical chart review of all brain donors for family history which allowed us to identify this case. We thank Dr. Aaron Day-Williams for oversight of sequencing analysis of the 188-gene panel. The study was financially supported by philanthropic donations to the Parkinson’s Institute and Clinical Center and sequencing analysis was supported by Biogen Idec.