
Abstract
Background:
NEFL encodes for the neurofilament light chain protein. Pathogenic variants in NEFL cause demyelinating, axonal and intermediate forms of Charcot-Marie-Tooth disease (CMT) which present with a varying degree of severity and somatic mutations have not been described yet. Currently, 34 different CMT-causing pathogenic variants in NEFL in 174 patients have been reported. Muscular involvement was also described in CMT2E patients mostly as a secondary effect. Also, there are a few descriptions of a primary muscle vulnerability upon pathogenic NEFL variants.
Objectives:
To expand the current knowledge on the genetic landscape, clinical presentation and muscle involvement in NEFL-related neurological diseases by retrospective case study and literature review.
Methods:
We applied in-depth phenotyping of new and already reported cases, molecular genetic testing, light-, electron- and Coherent Anti-Stokes Raman Scattering-microscopic studies and proteomic profiling in addition to in silico modelling of NEFL-variants.
Results:
We report on a boy with a muscular phenotype (weakness, myalgia and cramps, Z-band alterations and mini-cores in some myofibers) associated with the heterozygous p.(Phe104Val) NEFL-variant, which was previously described in a neuropathy case. Skeletal muscle proteomics findings indicated affection of cytoskeletal proteins. Moreover, we report on two further neuropathic patients (16 years old girl and her father) both carrying the heterozygous p.(Pro8Ser) variant, which has been identified as 15% somatic mosaic in the father. While the daughter presented with altered neurophysiology,neurogenic clump feet and gait disturbances, the father showed clinically only feet deformities. As missense variants affecting proline at amino acid position 8 are leading to neuropathic manifestations of different severities, in silico modelling of these different amino acid substitutions indicated variable pathogenic impact correlating with disease onset.
Conclusions:
Our findings provide new morphological and biochemical insights into the vulnerability of denervated muscle (upon NEFL-associated neuropathy) as well as novel genetic findings expanding the current knowledge on NEFL-related neuromuscular phenotypes and their clinical manifestations. Along this line, our data show that even subtle expression of somatic NEFL variants can lead to neuromuscular symptoms.
Keywords
INTRODUCTION
Neurofilament light chain (NEFL) protein is one of the neurofilament-core-subunits, forming heterodimers. Neurofilaments (NFL) provide structural stability to neurons. They are essential for the radial growth of axons during development, the expansion and maintenance of axonal caliber, and the transmission of electrical impulses on axons [1, 2]. NEFL proteins are subunits of neurofilaments in both the central and the peripheral nervous system and as such, dysfunction of the NEFL protein could give rise to pathology in either of the two parts of the nervous system [3]. Toxic neurofilament-accumulation is a hallmark of many neurodegenerative disorders like autoimmune-mediated motor neuron injury [4], amyotrophic lateral sclerosis (ALS) [5], Charcot-Marie-Tooth disease, 5q-related spinal muscular atrophy (SMA), spastic paraplegia, Alzheimer’s disease and Parkinson’s disease [6].
Charcot-Marie-Tooth (CMT) has been classified electrophysiologically into three major phenotypes: predominantly demyelinating forms (CMT1), predominantly axonal forms (CMT2) and dominant-intermediate forms [2]. Pathogenic NEFL variants may cause demyelinating (CMT1F), axonal (CMT2E) and dominant intermediate forms (DICMTG). Hereby, the disease-associated variants are located throughout the functional domains of the protein, which associates with subunits of the “medium” (NEFM) and the “heavy” (NEFH) protein versions to form coiled-coil dimers resulting in the build-up of 10 nm sized intermediate filaments [11].
Currently, more than 30 different CMT-causing pathogenic variants in NEFL have been reported, which embrace the head, rod, and tail domains of the protein. Most of the mutations are dominant missense variants functioning through a gain-of-function mechanism by perturbing neurofilament assembly and organelle transport in axons [12]. Only three autosomal recessive nonsense mutations were reported with associated severe, early-onset phenotypes [13–15]. However, current phenotype-genotype correlations suggest a loss-of-function mechanism due to the lack of NF networks for bi-allelic variants whereas the impact of the different dominant variants on respective phenotypical manifestations are still not completely understood [2, 17]. Further studies in a primary motor neuron culture model indicated that mitochondrial dysfunction contributes to the pathogenesis of CMT2E [18]. This might be one of the reasons for the presence of additional, non-neuropathic symptoms including hereditary spastic paraplegia, progressive spasticity, intellectual impairment and hearing loss in some of the patients [16, 19–21]. Muscular involvement is described as a clinical feature in CMT2E patients mostly recognized as a secondary effect but overlapping myopathic findings and clinical features of a congenital myopathy were described in some cases [17]. Increased creatine kinase (CK) levels and additional myopathic changes including myofiber size variation, rounding of fibers, ring fibers, and nemaline bodies in muscle biopsies of CMT2E patients are a further hint towards a concomitant muscular effect of pathogenic NEFL variants [22]. These descriptions accord with the results of functional studies obtained in a porcine model of muscle development, showing that expression of NEFL plays a significant role in embryonic morphogenesis and skeletal muscle development [23].
Interestingly, pathogenic variants affecting other genes (such as LMNA, DNM2 and BAG3) are associated with the manifestation of diseases across the neuromuscular axis and may cause myopathy or neuropathy [24, 25]. This phenomenon may be related to disruption of different tissue-specific functions of the respective proteins [26]. However, studies focusing on NEFL-based myopathological changes in human muscle tissue are still rare.
PATIENTS, MATERIALS AND METHODS
Patients
Patients included in this study were phenotyped in the Department of Pediatric Neurology of the University Hospital of Essen (Duisburg-Essen University). Written informed consent for clinical description, genetic studies, and utilization of the muscle biopsy for research purposes was obtained from the patients’ parents. The study was conducted according to the guidelines of the Declaration of Helsinki and approved by the Ethics Committee of University of Duisburg-Essen (19-9011-BO).
Molecular genetic studies
Genomic DNA was obtained from total peripheral blood samples using routine extraction methods. Next-generation sequencing (gene panel analysis including 1.678 genes with relevance in clinical diagnostics, custom-made target enrichment, Agilent Sure Select) was carried out on an Illumina NextSeq 500 system (Illumina, San Diego, CA) as 150 bp paired-end runs using v2.0 SBS chemistry. Sequencing reads were aligned to the human reference genome (GRCh37/hg19) using BWA (v0.7. 13-r1126) with standard parameters. Statistics on coverage and sequencing depth on the clinical targeted regions (i.e. RefSeq coding exons and +/–5 intronic region) were calculated with a custom script. SNV and INDEL calling on the nuclear genes was conducted using SAMtools (v1.3.1) with subsequent coverage and quality dependent filter steps. Variant annotation was performed with snpEff (v4.2) and Alamut-Batch (v1.4.4). Only variants (SNVs/small INDELs) in the coding and flanking intronic regions (±50 bp) were evaluated. BayesDel (https://fenglab.chpc.utah.edu/BayesDel/BayesDel.html) and the thresholds published by Pejaver et al. [27] were used for bioinformatic predictions. Variants were classified according to the ACMG (American College of Medical Genetics and genomics) guidelines taking the current recommendations from ClinGen's Sequence Variant Interpretation Working Group (SVI) into account [16, 28] and classifying variants with the 5-tier classification system: class 5 (pathogenic), class 4 (likely pathogenic), class 3 (uncertain variants or variants of unknown significance, VUS), class 2 (likely not pathogenic) and class 1 (not pathogenic).
In silico modelling of amino acid substitutions affecting Pro 8
ColabFold version 1.5.2 was employed using the AlphaFold2_mmseqs2 Jupyter notebook to predict the three-dimensional structures of the wild-type NEFL protein (UniProt accession: P07196) and five missense NEFL protein variants [29]. Each three-dimensional structure was predicted alongside pLDDT score, which is a per-residue confidence metric between 1–100 generated by AlphaFold2 [30]. PyMOL was then used to individually superimpose the wild-type NEFL protein model onto each of the missense protein variant models using the “super” command. The “super” command also computes a root-mean-square deviation (rmsd) score for each superimposition, representing structural similarity between the two models [31]. Finally, the predicted protein models were uploaded to PremPS, a web server that can be used to predict the effects of missense mutations on protein stability, measured by unfolding free energy change (ΔΔG) [32]. The five NEFL missense mutations were specified to the PremPS server, and the changes in non-covalent interactions between wild-type NEFL and each missense variant were modelled, exclusively focusing on non-covalent interactions between the mutated amino acid residue and its adjacent residues. A ΔΔG value (kcal/mol) was estimated for each mutation as an indicator of protein stability change as a result of the mutation, with positive and negative ΔΔG values indicating mutations that either destabilize or stabilize the protein structure, respectively [32].
Biopsy work-up
Muscle biopsy specimen investigated in this study (from patient 1) has initially been collected for diagnostic purposes including histology, enzyme histochemistry, immunofluorescence and immunohistochemical investigations. Serial cryosections (7μm) of transversely-oriented muscle blocks were stained according to standard procedures with hematoxylin and eosin (H&E), Gömöri trichrome (GT), oil red, COX-SDH and SDH and nicotinamide adenine dinucleotide tetrazolium reductase (NADH-TR) [33]. Microscopy was performed using a Zeiss Axioplan epifluorescence microscope and a Zeiss Axio Cam ICc 1.
Glutaraldehyde-fixed specimens were processed for ultrastructural examination by standard procedures. The tissue was post-fixed in 1% osmium tetroxide and embedded in Epon 812. Semithin sections for light microscopy were stained with toluidine blue. Ultrathin sections were contrasted with uranyl acetate and lead citrate and examined using a Philips CM10 transmission electron microscope as described [34].
Coherent anti-Stokes Raman scattering microscopy (CARS)
From a cyro-preserved muscle biopsy specimen, 5μm sections were prepared and used for coherent anti-Stokes Raman scattering (CARS) as well as for second harmonic generation (SHG) microscopy. Samples were dried in a nitrogen stream and required no further sample preparation.
For the CARS/SHG measurements, a modified Leica TCS SP 8 CARS was used with an APE picoEmerald as the laser source (as described previously, [35]). CARS and SHG signals were detected simultaneously in backward (Epi; E) and in forward (F) direction. For the imaging, we used a 40× water immersion objective lens (IRAPO 40×/1.10 WATER). For imaging and acquisition of CARS spectra, a field of 291×291μm (2048×2048 pixels) was measured on the samples. By tuning the pump laser from 804.0 nm to 826.4 nm with a step size of 0.7 nm, a pixel dwell time of approximately 10μs and averaging of two images, we acquired the FCARS spectra.
Statistical evaluation of muscle fibre calibres
Length and width of fully imaged muscle fibres were manually measured from merged FCARS/ESHG images utilizing the Leica software Las X. The muscle fibre calibre was averaged from the two lengths. For muscle fibres in longitudinal orientation, width of the fibres was determined as the calibre. For this purpose, a total of 6 images of the patient and 8 images of the disease control were analysed.
Acquisition of the spectra of the feature findings
The spectral CARS/SHG measurements were manually screened for conspicuous features. These were grouped according to their appearance (see results section). The CARS spectra in each group were first normalized and then averaged. For comparison, inconspicuous areas neighbouring the features (BG) were handled alike. A total of 194 spectra were used for the spectra presented in the results section: pBG: 45, pProt: 45, dcBG: 69, dcProt: 11, dcLip: 24, cBG: 135.
Proteomic profiling
Proteomic profiling of whole muscle protein extracts was carried out as described previously in a data-independent-acquisition mode [36].
RESULTS
Clinical findings
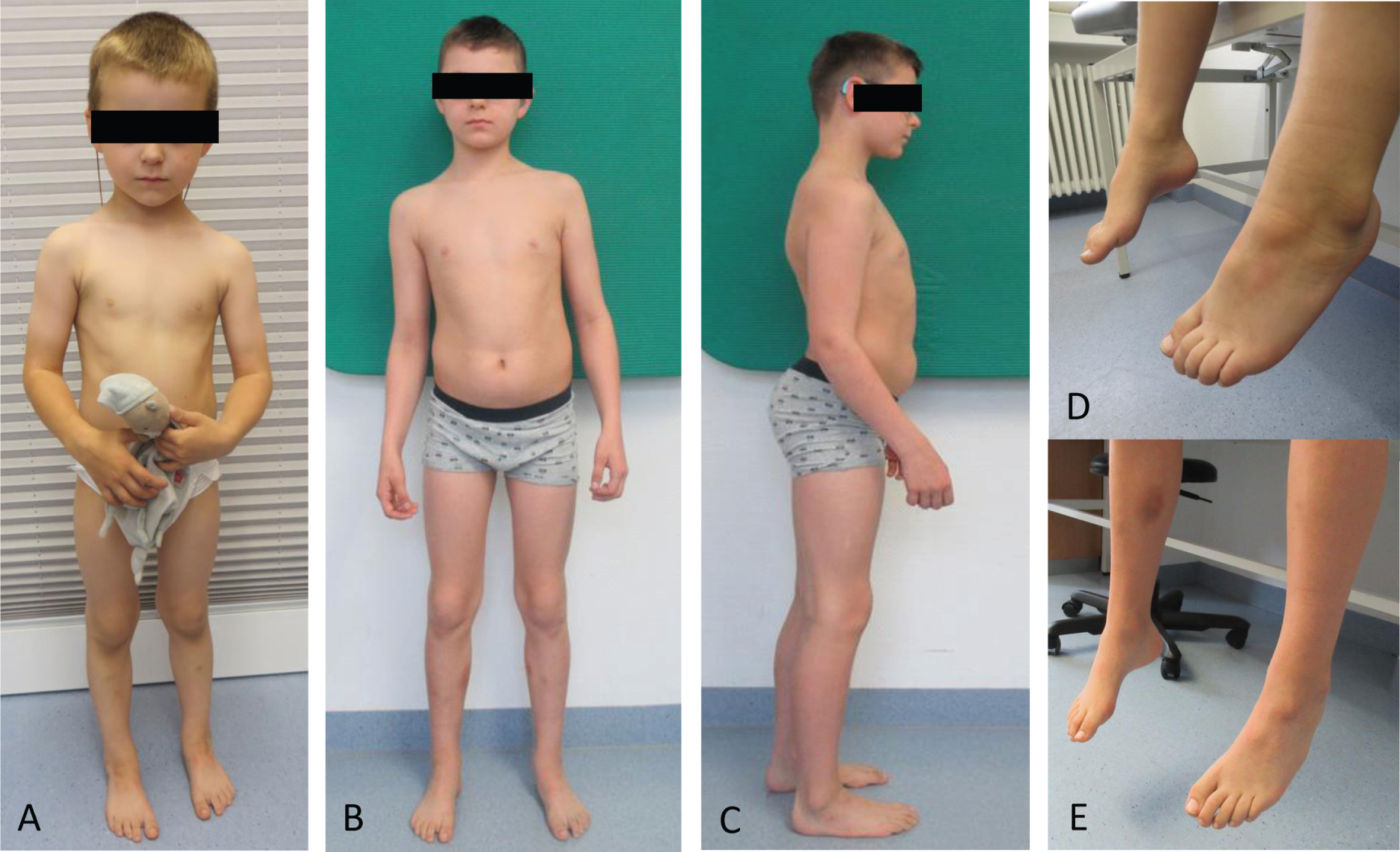
Clinical findings in one NEFL-patient. Patient 1 at the age of 3 years (
Summary of clinical, molecular genetic and in silico prediction based findings in patient harbouring amino acid substitutions affection position 8 (modified based on Jordanova et al 2003 [38]). Overview of the clinical and neurophysiological data from patients harbouring mutations in codon 8 of NEFL reported in the literature including our patients [14, 57]. In silico modelling results estimated either stabilising or destabilising ΔΔG values for the five NEFL variants. NCV –nerve conduction velocity, LE –lower extremity, UE –upper extremity, ND –not determined, ATR –Achilles tendon reflex
Molecular genetic findings
In silico modelling of amino acid substitutions affecting proline at position 8
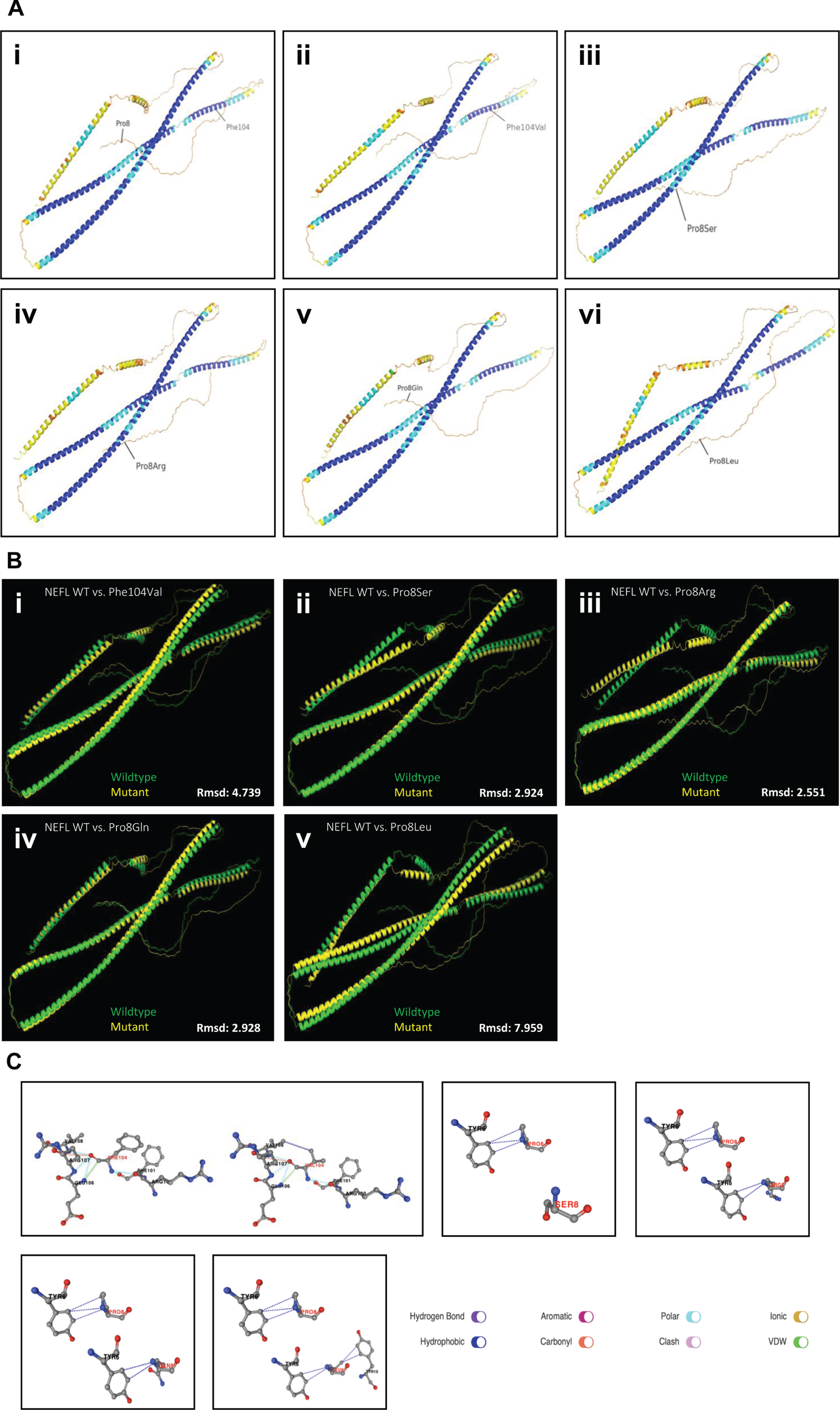
To address the impact of the nature of amino acid substitutions on the manifestation of clinical symptoms in terms of severity of phenotypes associated with different missense variants affecting amino acid position 8, Alphafold2-based in silico studies were carried out: the six NEFL protein structures were predicted with pLDDT scores ranging from 71.8–72.4 (Fig. 5A). When superimposed onto the wild-type NEFL protein model, the Phe104Val and Pro8Leu protein variants exhibited rmsd scores of 4.739 Å and 7.959 Å, respectively, which were the highest rmsd scores among the modelled variants (Fig. 5B & Table 1). On the other hand, Pro8Ser, Pro8Gln, and Pro8Arg exhibited rmsd scores between 2.5 Å and 3.0 Å when superimposed onto wild-type NEFL (Fig. 5B & Table 1). Identical structures are indicated by a rmsd of 0 Å, a rmsd up to 2.0 Å reflects good structural similarity, and a rmsd of 3.0 Å and above indicates low similarity [40–42]
PremPS predicted that all five variants alter the hydrophobic interactions between the mutated and its adjacent residues (Fig. 5C). The p.(Phe104Val) variant introduces a new hydrophobic interaction between Val104 and Val108. The Pro8Ser variant lacks all three hydrophobic interactions between Pro8 and Tyr6 that are present in wild-type NEFL. Pro8Arg leads to the loss of a hydrophobic interaction between Arg8 and Tyr6, and p.(Pro8Gln) leads to the loss of a hydrophobic interaction between Gln8 and Tyr6. Lastly, the p.(Pro8Leu) mutation causes one hydrophobic interaction to be lost between Leu8 and Tyr6 while also introducing two new hydrophobic interactions between Leu8 and Tyr10. All changes in hydrophobic interactions were associated with a ΔΔG value (kcal/mol) (Table 1). PremPS estimated either stabilising or destabilising ΔΔG values for the five NEFL variants. Both stabilising and destabilising ΔΔG values have been associated with deleterious mutations. The Pro8Leu variant showed a ΔΔG of –0.35 kcal/mol compared to wild-type NEFL, which was the highest magnitude ΔΔG value among the variants tested. Thus, our in silico predictions underline the pathogenic character of the amino acid substitution identified in our NEFL-patients and moreover demonstrate that missense variants affecting the same amino acid substitution may have diverging deleterious effects in terms of irregular stabilization or destabilization of NEFL.
Histological and electron microscopic findings on NEFL-mutant muscle
Histological examination did not reveal striking myopathic changes in the patient 1’s muscle biopsy (H&E staining shown in Fig. 2A) except the indication of altered mitochondrial distribution based on NADH-TR staining (Fig. 2B). However, ultra-structural studies utilizing electron microscopy unraveled focal alterations of sarcomere pattern with streaming of Z-discs and incidental pronounced Z-disc disruption as minicore-like lesions in addition to small cytoplasmic bodies (Fig. 2C & D). No larger autophagic vacuoles, or tubular aggregates were identified. Mitochondria presented with normal size and shape and no ultrastructural abnormalities of nuclei were found (data not shown).
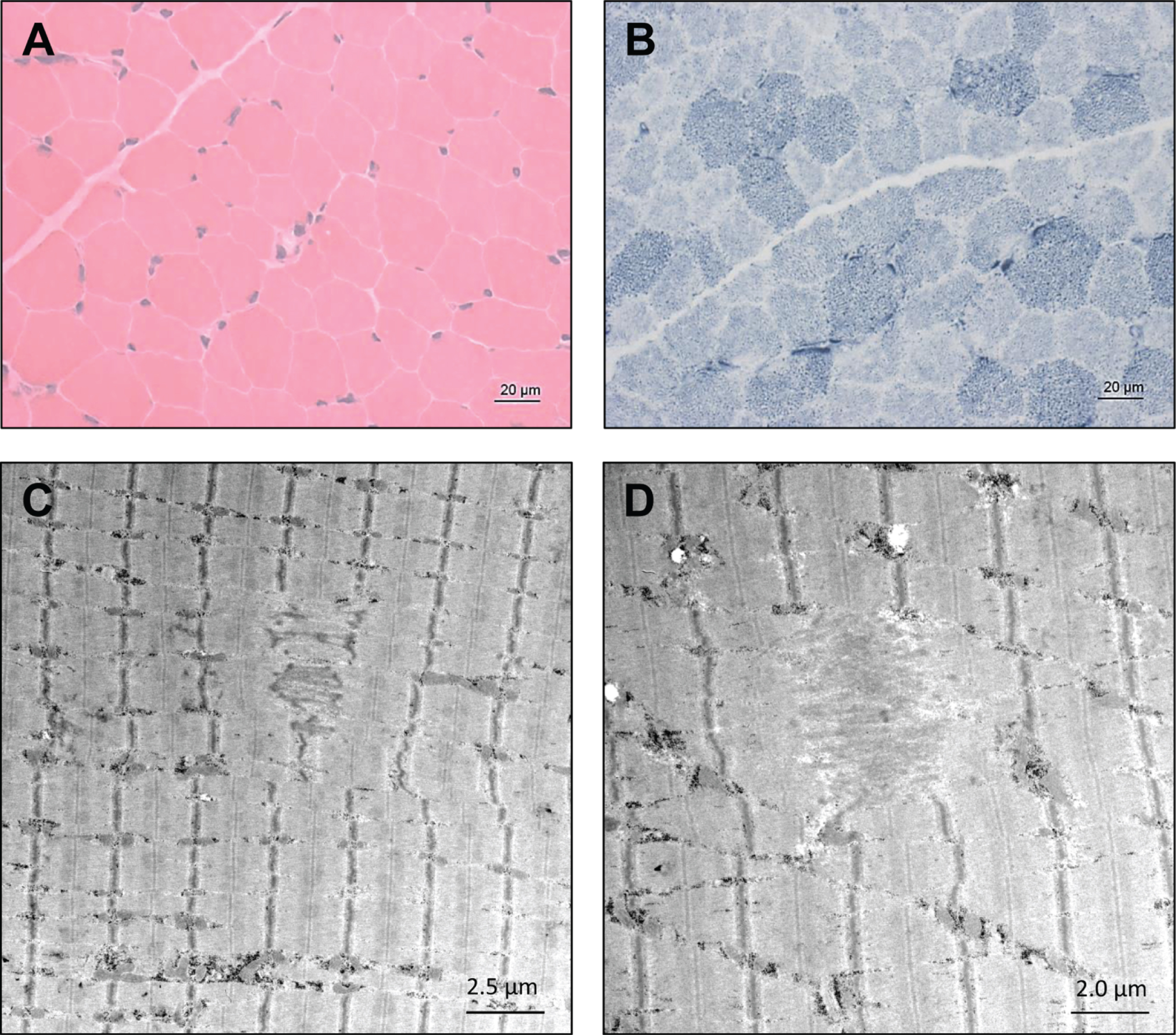
Microscopic findings in NEFL-mutant muscle. Haematoxylin & Eosin (H&E) staining did not reveal striking myopathic changes (
Coherent anti-Stokes Raman scattering microscopy (CARS)
CARS offers the unique opportunity to investigate protein and lipid imbalances in an unbiased manner by making use of a minimal amount of material and is thus a valuable analytical technique in the investigation of pathological features in biopsy material such as muscle biopsy specimens. Thus, we applied this label-free technique to obtain further unbiased insights into the muscle cell vulnerability underlying the pathogenic NEFL-variant. To locate spectroscopic features in the muscle biopsy samples (NEFL-patient 1 and controls), first the spectrum of the fibre background (areas in which no features) was defined, and a mean spectrum was calculated. The fibre background of NEFL-patient 1 (Fig. 3A, pBG) shows a lower signal intensity at 2921 cm–1 than at 2868 cm–1. The signal at 2921 cm–1 is characteristic for proteinogenic content [43, 44]. The standard peak of ordered lipids [45] at 2889 cm–1 seems to be shifted to 2868 cm–1. In contrast, in subsarcolemmal regions of NEFL-mutant muscle fibres, the intensity at 2921 cm–1 and 2868 cm–1 is comparable (Fig. 3A, pProt). For CARS A, a higher intensity for 2921 cm–1 based spectra can be observed in the subsarcolemmal regions compared to the fibre background. In the light of the above-mentioned aspects, we concluded based on these data that in NEFL-mutant muscle demonstrates an increased protein content (mirrored by increased signal at 2921 cm–1) is present in some regions of fibers.
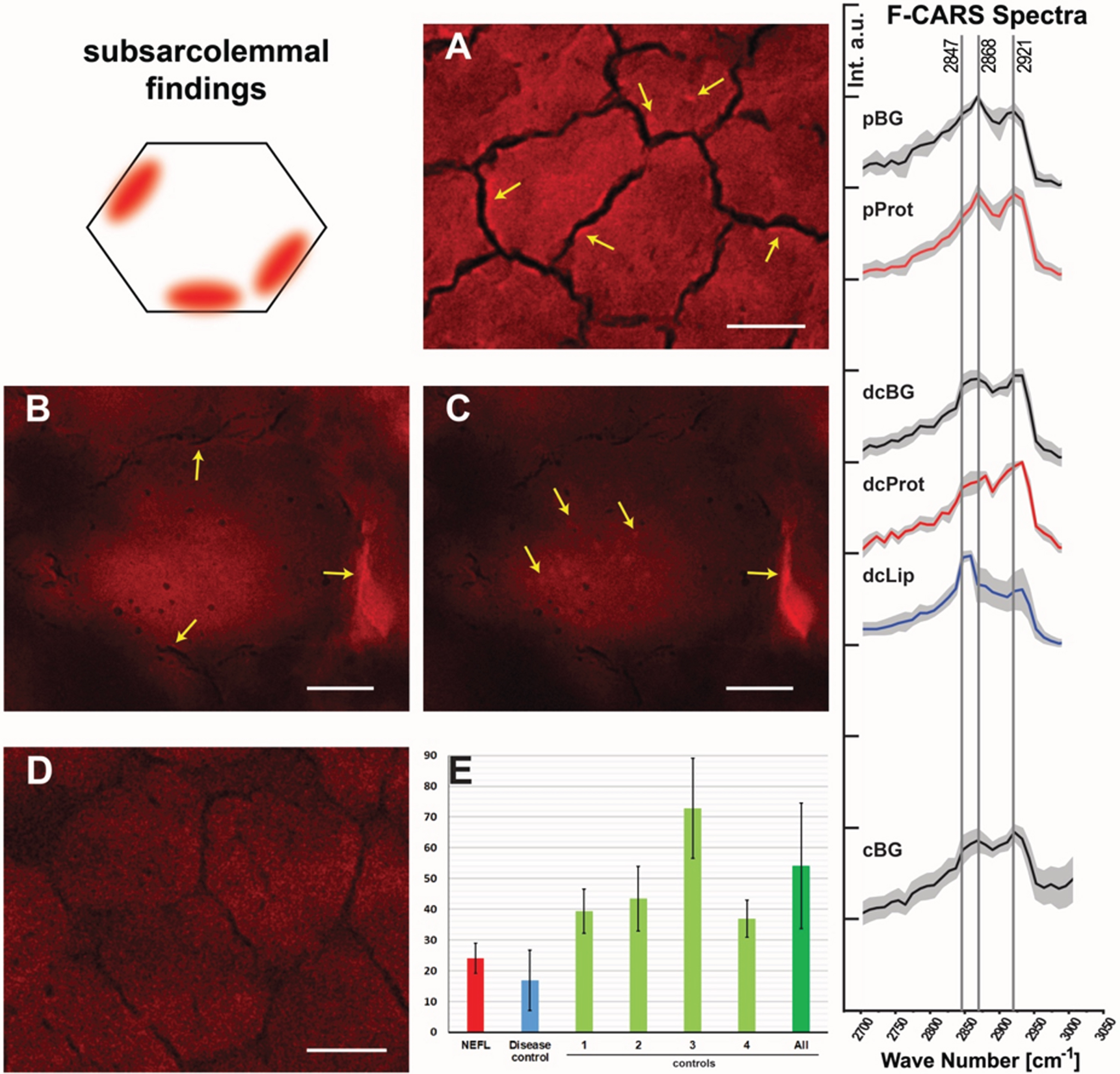
Spectroscopic findings in NEFL-mutant muscle obtained by F-CARS measurements. Protein-containing regions in muscle fibres of NEFL-patient 1 at 2921 cm–1 (marked with arrows; scale bar 10μm) (
Spectral analysis of the disease control (neurogenic muscular atrophy in the biopsy of an age-matched child) revealed differences in comparison to the NEFL-patient 1 (Fig. 3B & C). The fibre background (dcBG) shows a slightly higher intensity at 2921 cm–1 than at 2868 cm–1. Thus, the spectrum is similar to that of the controls (Fig. 3D, cBG). Also, less pronounced subsarcolemmal spots with significantly increased signal at 2921 cm–1 were found in the disease control (Fig. 3, dcProt), but regions in and between fibres were also identified with high signal at 2847 cm–1 (Fig. 3C, dcLip). Especially disordered lipids show a strong signal at 2847 cm–1. No lipid features were found, neither in controls nor in the NEFL-patient.
To investigate muscle fibre calibre, we measured the average of muscle cells in the biopsy derived from NEFL-patient 1 in comparison to a disease control (denervation atrophy) and normal controls: while the averaged muscle fibre calibre in the NEFL-patient 1’s muscle biopsy is 24.01μm±4.94μm (721 fibres), a calibre of 54.09μm±20.33μm (1027 fibres) was detected in the normal controls and 16.97μm±9.78μm (782 fibres) in the disease control (Fig. 3E). Thus, our CARS-based investigations also enabled to demonstrate smaller muscle fibre calibre in NEFL-patient 1, a microscopic finding which accords with the assumption of neuropathy-related denervation.
Proteomic profiling of NEFL-mutant muscle derived from patient 1
To identify the proteins (reflecting molecular processes) underlying the pathogenic NEFL variant in skeletal muscle, we performed label-free untargeted proteomic profiling on quadriceps muscles derived from patient 1 in comparison to three healthy age- and gender-matched controls (Fig. 4A). By measuring 5777 unique peptides (Fig. 4B), we identified 58 proteins with an increased abundance of more than 1.3-fold whereas 60 proteins were identified with a decrease of a minimum of 0.7-fold (Fig. 4C & Table 2). These 118 altered proteins were quantified based on at least two unique peptides and significantly dysregulated with a p-ANOVA≤0.05 (Table 2). All functional information about these dysregulated proteins listed in Table 1 was extracted from uniprot (www.uniprot.org, accessed on 10th of March 2023). We performed GO term analysis focussing on the up- and down-regulated proteins separately (Fig. 4D): within the group of upregulated proteins, the most altered biological processes include organization of the extracellular matrix including collagen fibril organization (indicative for fibrotic processes), activity of complex I of mitochondrial respiratory chain and metabolic processes of reactive oxygen species and carbohydrates, tricarboxylic acid cycle, axogenesis including axon extension involved in axon guidance, chaperone-mediated protein folding and muscle contraction (Fig. 4B). Molecular functions affected by increased proteins contain oxidoreductase activity including NADH dehydrogenase activity and electron carrier activity, extracellular matrix structural constituent and matrix binding, double-stranded RNA binding, activities of superoxide dismutase and thioester hydrolase, binding of proteins to chaperones (to warrant proper folding) and muscle alpha-actinin protein binding (Fig. 4D). In accordance with affected biological processes and molecular functions, affected cellular compartments include the extracellular matrix, mitochondria, lumen of Golgi apparatus and lysosomes, peroxisomes, nuclear chromatin, sarcolemma including membrane rafts, actin cytoskeleton and the myelin sheath (Fig. 4D). A GO-term-based pathway analysis of down regulated proteins revealed that a significant proportion of these proteins affect different biological processes including glycolysis, tricarboxylic acid cycle, mitochondrial acetyl-CoA biosynthetic processes, creatine metabolic processes, muscle organ development and functions such as muscle contraction and myofibril assembly. Molecular functions affected by decreased protein abundances contain structural constituent of muscle and cytoskeleton (including binding of cytoskeletal proteins such as titin and tropomyosin), ion channel binding as well as activities of structural proteins and pyruvate dehydrogenases (Fig. 4D). In line with the affected biological processes and molecular functions decreased proteins impact on myelin sheath, extracellular matrix, myonuclei, mitochondria and the sarcomeric system (Z-disc, I and M bands, actin and thin filaments) (Fig. 4D).
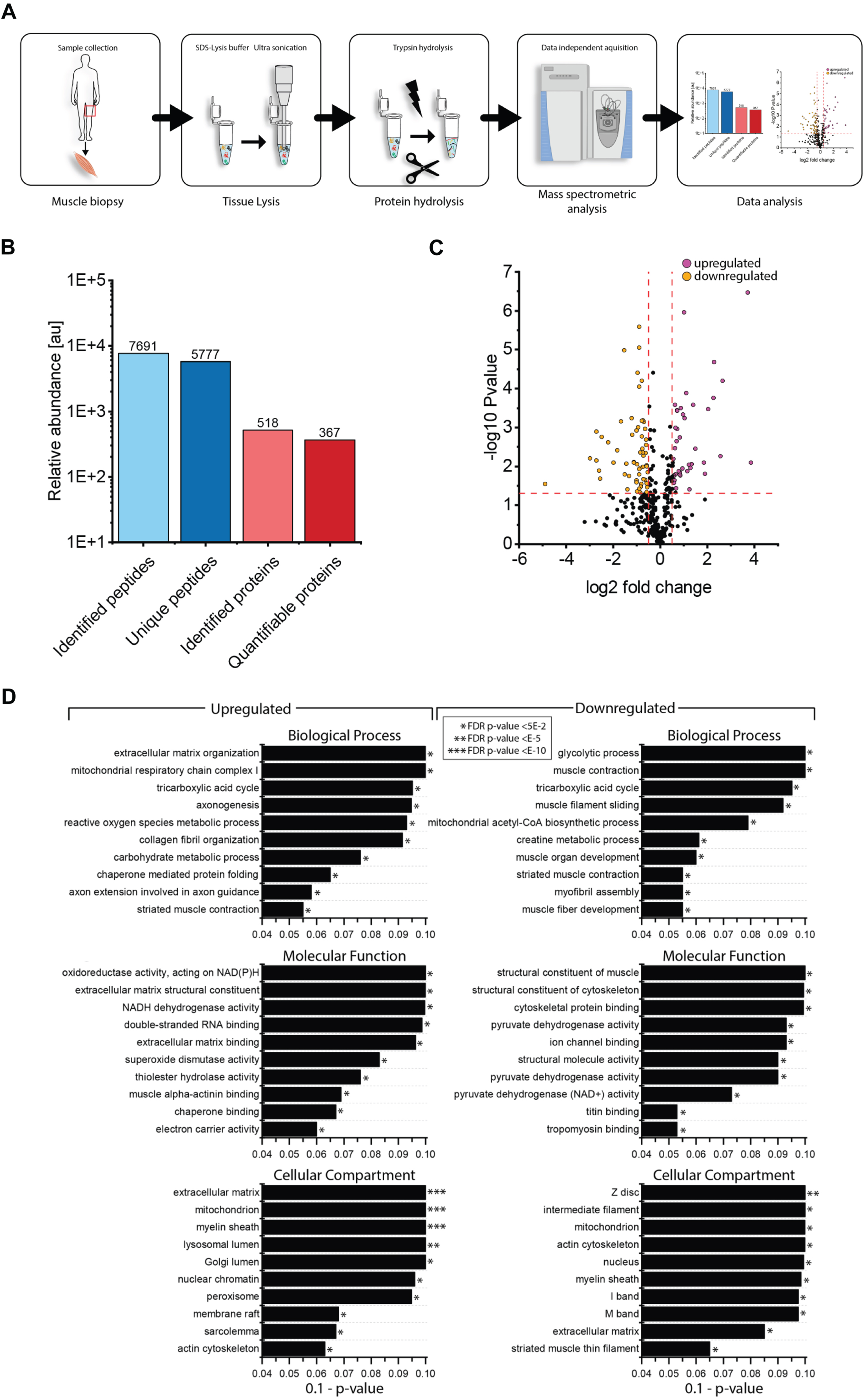
Proteomic profiling of NEFL-mutant muscle. (
In silico studies on amino acid substitutions affecting NEFL. (
List of statistically significant dysregulated proteins including number of unique peptides identified for each protein
DISCUSSION
Pathogenic variants in NEFL have been frequently linked to manifestation of different subtypes of CMT neuropathy, CMT1F and CMT2E and an intermediate form [46]. Most pathogenic NEFL variants are dominant missense variants functioning through a gain-of-function mechanism in which amino acid substitutions within the corresponding protein disrupt neurofilament assembly and thus impact organelle transport within cells of the peripheral nervous system. All reported homozygous nonsense variants follow a recessive mode of inheritance [13–15]. Besides NEFL serves as a biomarker in cerebrospinal fluid of neurodegenerative diseases such as Amyotrophic Lateral Sclerosis and 5q-related spinal muscular atrophy. Furthermore, a transgenic mouse model of a mutant neurofilament light chain p.(Leu394Pro) shows loss of ventral horn motor neurons and denervation atrophy of the skeletal muscle suggesting that NEFL may also be a candidate gene for motor neuron diseases [47]. These combined findings suggest a profound role of the protein in maintenance of the nervous system. However, studies aiming to decipher molecular processes associated with porcine intrauterine muscle cell development revealed that altered Nefl expression is associated with the process of primary to secondary myofibres formation [23] in turn suggesting a functional role of the neurofilament light chain protein in muscle. Of note, based on cases presenting with pathogenic variants in NEFL associated with the manifestation of primary myopathic features, there is growing evidence for a functional role of this intermediate filament also in skeletal muscle: one family with 4 affected members (mother and her three sons all carrying the c.1261C > T variant) presented with different pathomorphological findings in the muscle biopsy: in addition to neuropathic features (including type II fiber predominance, grouped fiber atrophy, and small angular fibers), pathomorphological findings such as Nemaline bodies indicative for a primary vulnerability of muscle cells (myopathy) were identified [17]. The NCV and muscle biopsy findings (including determination of muscle fibre calibre by CARS) in our NEFL-patient 1 were compatible with neurogenic process. However, neuropathic changes occasionally can be seen in a chronic myopathic process with GFPT1- and BICD2-related neuropathologies being prominent examples [48, 49]. In a previous study, we reported on a family presenting with the c.1186G > A; p.(Glu396Lys) variant not only associated with mixed axonal and demyelinating neuropathy but also with considerably elevated CK levels in all affected adults of the family as well as pronounced myopathic changes in skeletal muscle biopsies (increased number of internalized myonuclei, myophagic reaction indicating muscle fibre break down) pointing towards an accompanying muscle involvement as a primary target [22]. These data already suggest a clinical variability associated with pathogenic NEFL variants and indeed recently a novel NEFL variant (c.1319C > T) was linked to the manifestation of a mild form of CMT2E associated with liability to pressure palsy (HNPP) [50]. Moreover, another recent study described cohort of NEFL patients presenting with an intermediate form of neuropathy complicated a range of symptoms including cardiac conduction abnormalities [51].
Important to note, our patient 1 presented with muscle cramps and muscular stiffness as first clinical symptoms, indicative –in combination with mildly elevated CK levels –for primary muscular involvement in terms of myopathy impacting on diagnostic work-up (including microscopic studies on muscle biopsy). Hence, here, we link the c.310T > G; p.(Phe104Val) variant to the manifestation of a mild neuropathy associated with muscle symptoms defined by cramps, mildly elevated CK level and focal myofibrillar alterations with minicore-like disintegration of sarcomeric structures. In this context it is important to note that different degenerative changes in Z-disc integrity were already linked to neuropathies defined by axonal pathology with decrease of neurofilaments and microtubules in myelinated and unmyelinated nerves [52]. Thus, that ultrastructural pathology of sarcomeres in our patient may result from denervation, as already hypothesized by Agrawal and colleagues describing a family with a primary neuropathic phenotype in the context of the extremely low expression of NEFL in skeletal musculature [17]. However, further mycological studies for instance on a suitable animal model are crucial to draw final conclusions.
CARS microscopic studies on the muscle biopsy specimen derived from our patient 1 in comparison to non-disease controls as well as a disease control (muscle denervation) revealed presence of subsarcolemmal protein enrichment and investigation of the proteomic signature unravelled dysregulation of proteins according with the ultra-structural findings: a variety of cytoskeletal proteins were dysregulated including FLNc as a protein for which pathogenic variants have been linked to a muscle disease called myofibrillar myopathy, which is characterized by disintegration of sarcomeric structures. Increased expression of chaperones (needed for protein folding) was also identified in patient 1’s quadriceps muscle. Further biochemical studies –such as proteomics - on denervated muscle are crucial to elucidate the impact of FLNc (and other structural proteins) as well as of chaperones on disintegration of sarcomeric structures resulting from perturbed neuromuscular transmission.
Regarding the broad clinical manifestation of pathogenic NEFL variants, it is important to note that recently, also central nervous system (CNS) affection including abnormal MRI findings, dementia and dysarthria have been described [53–55]. Already Kim and colleagues postulated that NEFL plays also a role in the CNS and supported this assumption by the finding of multiple inclusions in cell bodies and proximal axons of the spinal cord, cerebellum, pons, and cerebral cortex in Nefl-mouse model (heterozygous p.(Asn98Ser) variant) [53]. Moreover, in NEFL-patients carrying the p.(Glu90Lys) and p.(Asn98Ser) amino acid substitutions, intellectual impairment was present [16]. Thus, we consider the intellectual disability present in our patient 1 also as a part of CNS involvement. However, his cranial MRI at the age of two years was normal.
One might speculate that the pathogenicity of respective amino acid substitutions or expression of modifying factors contribute to the phenotypical manifestation and thus broad neurological/neuromuscular spectrum associated with pathogenic variants in NEFL. Indeed, further functional studies on patient derived material are needed to prove this assumption. Another explanation for clinical (intra-familiar) variability is provided by our familiar cases presented in this study: while patient 2 presents a neuropathy-phenotype (NCV at the age of 16 years revealed demyelinating pattern) with slow progressive muscular weakness, mild peripheral ataxia and reduced walking distance and pes cavus, her father had a non-progressive pes cavus with normal muscle strength, absent ATR and minimal neuropathic pattern as observed by the results of NCV measurement. Of note, while molecular genetic studies on DNA derived from the daughter clearly revealed the c.22C > T p.(Pro8Ser) variant, the same variant was detected only in 15% of reads indicative for mosaicism. Interestingly, three other amino acid substitutions affecting the same position (p.(Pro8Arg), p.(Pro8Gln) & p.(Pro8Leu)) have been described before [38]. A clinical comparison revealed that these variants are associated with both, a more (p.(Pro8Leu) & p.(Pro8Gln)) as well as a less severe (p.(Pro8Arg)) phenotype than observed in our patient. Intra-familiar variability has been described in the context of clinical manifestation of pathogenic NEFL variants and should thus carefully be taken into consideration in the delineation of the attempts of genotype-phenotype correlations. Along this line, a muscular phenotype segregating in a family with four affected members included different grades of severity of clinical manifestation [17]. One might speculate that the presence of modifiers may explain the varying manifestation. However, comprehensive molecular studies on intrafamilial biosamples would be needed to systematically address this assumption. Nevertheless, it has been postulated that disease is not only caused by impairment of domain-specific functions of NEFL or neurofilaments but rather by general perturbations in protein folding or stability leading to pathological changes in neurofilament subunit stoichiometry, or to protein aggregation and cellular stress [2, 53]. Along this line, more than 170 patients with pathogenic NEFL variants have been reported with broad age-range of the onset of the symptoms [2, 16] and it was postulated that pathogenic variants affecting the head domain lead to an earlier manifestation of the phenotype. Our in silico based modelling of missense variants affecting the amino acid proline at position 8 revealed a varying pathogenic impact of the respective amino acid substitutions either resulting in stabilization of destabilization of the protein. According to the clinical data available for our patient and in the literature one might speculate that stabilizing variants such as p.(Pro8Leu) and p.(Pro8Ser) are associated with an earlier disease manifestation. Of course, the current number of cases presenting with pathogenic variants affecting proline at position 8 of NEFL is too small to draw final conclusions and the possibility of other factors such as genetic modifiers also need to be considered. However, our combined clinical and molecular genetic findings support this assumption by introducing the p.(Pro8Ser) amino acid substitution as a novel NEFL variant extending the current molecular genetic landscape for this gene with an associated early clinical manifestation.
CONCLUSIONS
Based on our combined data, we expand the current molecular genetic landscape of NEFL-associated neuropathies by linking the c.22C > T; p.(Pro8Ser) variant to the manifestation of neuropathic features and along this line introduce the same variant in mosaicism in the mildly affected patient’s father. Hence, this is the first report of a pathogenic somatic NEFL variant. Given that different amino acid substitutions for position 8 were associated with diverging neuropathic manifestations, in silico modelling of these variants was performed and results suggest a link between protein misfolding and age of onset.
Moreover, our report on a boy with a NEFL variant known to cause neuropathy but also suffering from muscular symptoms (supported by microscopic and biochemical data) supports the concept that NEFL variants may have different clinical manifestations along the neuromuscular axis. Although, our data obtained in the latter patient imply additional muscle cell vulnerability upon the expression of mutant NEFL, the effect of denervation on morphological and biochemical integrity must be taken into consideration. Hence, further studies –for instance on human muscle organoids expressing mutant NEFL –would be needed to decipher the exact impact of the intermediate filament protein for proper muscle cell function.
AUTHOR CONTRIBUTIONS
AR, HK and ADM conceptualized and designed the study, drafted the first version of the manuscript, interpreted results and are principally responsible for the final content. USS and TR reviewed and revised the manuscript for important intellectual content. JW performed the electron microscopic studies. AH performed the proteomic studies. AC, AA, AL and AS analyzed data and interpreted results. In silico modelling was performed by HL and CC. All authors contributed to the final manuscript and agreed to be accountable for all aspects of the work.
Footnotes
ACKNOWLEDGMENTS
The authors are grateful to the families for cooperation and for the permission to publish the data. Four authors of this publication are members of the European Reference Network for Neuromuscular Diseases –Project ID N° 870177.
FUNDING
AR received funding from The French Muscular Dystrophy Association (AFM-Téléthon) (grant 21644). AR and USS also acknowledge funding in the framework of the NME-GPS project by the European Regional Development Fund (ERDF).
HL receives support from the Canadian Institutes of Health Research (CIHR) for Foundation Grant FDN-167281 (Precision Health for Neuromuscular Diseases), Transnational Team Grant ERT-174211 (ProDGNE) and Network Grant OR2-189333 (NMD4C), from the Canada Foundation for Innovation (CFI-JELF 38412), the Canada Research Chairs program (Canada Research Chair in Neuromuscular Genomics and Health, 950-232279), the European Commission (Grant # 101080249) and the Canada Research Coordinating Committee New Frontiers in Research Fund (NFRFG-2022-00033) for SIMPATHIC, and from the Government of Canada Canada First Research Excellence Fund (CFREF) for the Brain-Heart Interconnectome (CFREF-2022-00007).
CONFLICTS OF INTEREST
The authors have no conflict of interest to report.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon request.