
Abstract
Duchenne muscular dystrophy (DMD) is a severe form of muscular dystrophy (MD) that is characterized by early muscle wasting and lethal cardiorespiratory failure. While the mdx mouse is the most common model of DMD, it fails to replicate the severe loss of muscle mass and other complications observed in patients, in part due to the multiple rescue pathways found in mice. This led to several attempts at improving DMD animal models by interfering with these rescue pathways through double transgenic approaches, resulting in more severe phenotypes with mixed relevance to the human pathology. As a growing body of literature depicts DMD as a multi-system metabolic disease, improvements in mdx-based modeling of DMD may be achieved by modulating whole-body metabolism instead of muscle homeostasis. This review provides an overview of the established dual-transgenic approaches that exacerbate the mild mdx phenotype by primarily interfering with muscle homeostasis and highlights how advances in DMD modeling coincide with inducing whole-body metabolic changes. We focus on the DBA2/J strain-based D2.mdx mouse with heightened transforming growth factor (TGF)-β signaling and the dyslipidemic mdx/apolipoprotein E (mdx/ApoE) knock-out (KO) mouse, and summarize how these novel models emulate the metabolic changes observed in DMD.
ABBREVIATIONS
Apolipoprotein E Cytidine monophosphate-Nacetylneuraminic acid hydroxylase Dystrophin-associated glycoprotein complex Duchenne muscular dystrophy High-density lipoprotein Intermediate-density lipoprotein Knock-out Low-density lipoprotein Low-density lipoprotein receptor LDL receptor-related protein Latent TGF-β–binding protein 4 Muscular dystrophy Myoblast determination protein Total cholesterol Telomerase RNA component Triglycerides Transforming growth factor beta Very-low density lipoprotein
INTRODUCTION
Muscular dystrophies (MD) are a class of genetic diseases characterized by progressive muscle wasting and dysfunction. Duchenne MD (DMD), first described in the mid-1800 s [1, 2] by two unrelated investigators, is the most common form of MD; it is caused by the loss of functional dystrophin, a protein that plays a critical role as part of the dystrophin-associated glycoprotein complex (DGC), which anchors the intracellular actin cytoskeleton of a myofiber to the extracellular matrix [3]. Blunted dystrophin expression results in myofiber fragility, susceptibility to injury, loss of calcium and nitric oxide homeostasis [4, 5], muscle degeneration [6, 7] as well as early loss of ambulation and cardiorespiratory dysfunction in patients. Although steroid-based therapy is credited with extending the life of DMD children by lowering the rate of muscle function decline [8, 9], lifespan and quality of life in these patients remain greatly reduced. In the commonly used mdx mouse model of DMD, however, loss of dystrophin results in a much milder phenotype [10, 11] as early muscle damage is followed by much enhanced regeneration in most muscle tissues. Many have explored ways to improve DMD modeling by characterizing or generating laboratory dogs [12–14], pigs [15], rabbits [16], monkeys [17], cats [18] and rats [19] carrying dystrophin mutations. These generally present a more aggressive muscle phenotype and even terminal cardiorespiratory dysfunction, which are likely attributable to less efficient endogenous rescue mechanisms inherent to larger animals; however, the costs, complexity and ethical concerns associated with these more sophisticated models support the long-term objective of creating a more human DMD-like phenotype in mdx mice. Exacerbation of mdx disease severity has been reported on many occasions through advanced transgenic approaches that interfere with endogenous repair or compensatory pathways (briefly reviewed below) although with varying degree of relevance to the human condition.
An often-overlooked aspect of dystrophinopathies is the fact that they are increasingly considered multi-system metabolic diseases [20] independently of steroid treatment. DMD individuals show abnormalities in bone growth and density [21, 22], puberty [20], adipose tissue metabolism [23], vision [24], neurological functions [25], vascular changes and embolism [26, 27], even at preclinical stages [27], insulin sensitivity [28], liver complications [29], which suggest that a more holistic approach to DMD modeling may result in superior recapitulation of the complexity of human DMD. In this review, we compare the recent attempts at creating a human-relevant mdx mouse model and discuss how focusing on genetic modifiers and non-muscle abnormalities observed in DMD rather than muscle repair and regeneration correlate with major milestones in disease modeling improvements, such as the D2.mdx mouse [30] with heightened TGF-β activity [31] and the mdx/apolipoprotein E (mdx/ApoE) double knock-out (KO) mouse [32] with high total cholesterol (TC) [33], both of which mimic DMD metabolic abnormalities.
Early mdx-based double-mutant mice
Modeling of DMD in mice began with the original C57BL/10 mdx model of DMD, which harbors a naturally occurring point mutation in the Dmd gene that truncates and prevents significant expression of dystrophin [11]. Loss of dystrophin in mice results in very mild motor function deficits due to an early phase of muscle degeneration followed by intense hypertrophy and regeneration, with the exception of the diaphragm that undergoes more linear but non-terminal fibrosis [6, 35] New strains of mdx mice (mdx2cv,3cv,4cv,5cv) have been developed with a potent mutagen, and despite showing fewer dystrophin-positive “revertant” cells, they also show mild phenotypes similar to mdx mice with only minor increases in myocardial lesions, muscle fibrosis and inflammation [36, 37].
Advanced mdx-based double transgenic strains have been generated with the aim of studying the pathogenesis of DMD and better mimicking human DMD-associated muscle wasting, mostly by preventing compensatory rescue mechanisms typically observed in mice (Table 1; some reviewed in [38]). These advanced models of DMD rely on inactivation of utrophin, MyoD, α-dystrobrevin, dysferlin, α7-integrin, cytidine monophosphate-N-acetylneuraminic acid hydroxylase (CMAH), telomerase RNA component (TERC) and parvalbumin. Disease severity exacerbation can be achieved with these double transgenic mdx mice, however the relevance to DMD can be limited in part due to patients not harbouring the abnormalities induced by the second transgene mutation. Interfering with the finely regulated balance of muscle damage and regeneration in a non-DMD relevant fashion is therefore a major limitation of some of these models while the relevant mdx/TERC KO mouse [39] is generally not considered a metabolic humanization and is therefore only briefly mentioned herein.
Established dual-transgenic models of mdx severity exacerbation. Most of these advanced models of DMD interfere with muscle homeostasis pathways
The D2.mdx mouse and DMD modeling
The D2.mdx mouse represents one of the two major recent advances in mouse modeling of DMD that coincide with changes in whole-body metabolism. Indeed, in contrast to the above-mentioned mice that require sophisticated breeding strategies to introduce specific mutations to genes that directly regulate muscle homeostasis, D2.mdx animals were instead generated by backcrossing the original exon skipping-compatible C57BL10/ScSn mdx mouse, which harbors a dystrophin gene exon 23 mutation, to the DBA2/J mouse strain. DBA2/J is a sub-strain of the DBA background, one the oldest inbred mouse strain generated in the early 1900’s [40]. Compared to C57BL/10-mdx and DBA2/J control mice, the resulting D2.mdx mouse showed significant body mass reduction, exacerbated loss of hind limb muscle weight and increased muscle weakness, temporary reductions in left ventricular function, along with fewer myofibers [31]. D2.mdx mice also show blunted myofiber regeneration [41, 42] whereas muscle lesions exhibit increased fibrosis and fat accumulation with 10–25% of fibro-necrosis for the gastrocnemius, triceps brachii and quadriceps femoris muscles and approximately 30% for the diaphragm at 10 weeks of age [31, 43]. Also noteworthy is the exacerbated calcification of cardiac and skeletal muscles at an early age [42, 44], which as a whole supports the concepts that D2.mdx mice are more severe animal models of DMD and that disease-modifying genetic polymorphism may be present in certain strains.
Genome-wide scanning for locuses linked to DBA/2J-associated muscle wasting exacerbation in the context of γ-sarcoglycan deficiency identified polymorphism in the gene coding for latent TGF-β–binding protein 4 (LTBP4) [45], a protein that sequesters TGF-β and prevents its ability to activate TGF-β receptors. As the TGF-β family is often viewed as one of the most pleiotropic group of secreted cytokines with numerous isoforms, inhibitory complexes and receptors capable of regulating modulating cell division, fibrosis, tissue morphogenesis and regeneration [41, 46], amongst other biological activities, how exactly DBA2/J-associated loss of LTBP4 function affects key metabolic functions is poorly understood. The Ltbp4 gene encodes for 2 secreted ubiquitous LTBP4 isoforms that are part of the ECM glycoprotein and have a structural homology to fibrillins, and Ltbp4 gene knockout causes abnormal elastogenesis, lung development, cardiomyopathy, and colorectal cancer [47] whereas DBA2/J mice harbouring the LTBP4 gene mutation show high incidence of glaucoma [48], hearing loss and CD94-deficient NK cells [49, 50]. In addition to a having a truncated annexin A6 [51], a ubiquitously-expressed protein linked to calcium and membrane events, DBA2/J mice are also capable of little to no dietary cholesterol absorption and suffer from blood and hepatic cholesterol anomalies, lower blood glucose and smaller muscle mass than C57BL/10 or C57BL6/J mice [52–54]. As further discussed below, DBA2/J mice also show an unusually high degree of muscle calcification [55]. While the respective contribution of these multiple metabolic defects to the exacerbation of D2.mdx mice is currently unknown, high Ltbp4 expression has been reported in smooth and skeletal muscles [56–58], acting as a major DMD severity modifier through multiple single nucleotide polymorphisms (SNPs) [59] and there is some evidence to suggest that upregulation of TGF-β type II receptor is present in DMD patient muscle biopsies [60]. Overall, the improvements in DMD modeling enabled by the D2.mdx mouse are caused by a key DMD-associated disease modifying factor, in this case heightened TGF-β signaling, which coincides with major changes to whole-body metabolic homeostasis.
Despite the early enthusiasm generated by the D2.mdx mouse, ‘all that glitters is not gold’ as this new model of DMD also comes with caveats. While the D2.mdx mouse requires only a single transgenic approach to mutate the dystrophin gene and obtain a severe phenotype, DBA/2J is not as common as other strains, making mechanistic studies that often rely on knocking-out or overexpressing other genes more complex due to the need for congenicity. More worrying are the observations from long-term studies describing improvements –rather than worsening –over time of D2.mdx muscle calcification and despite continued muscle function decline, muscle histology reveals chronic stabilization of muscle lesions [44], in stark contrast to human DMD. Muscle pathology exacerbation is also likely more complex than simple LTBP4-deficient as D2.mdx-associated calcification is independent from TGF-β activity [61] and directly involve calcium homeostasis-modulating genes [44, 61]. Furthermore, it is unclear what other genetic factors present in the DBA/2J mouse may be contributing to the initially more severe mdx phenotype in the D2.mdx model which complicates its applicability to human DMD and its usefulness as a model for therapeutic development [62]. As a whole, whether D2.mdx mice can reliably improve DMD disease modeling remains to be determined.
DMD modeling and lipoprotein metabolism - the mdx/ApoE KO mouse
Another key milestone in humanizing the phenotype of mdx mice that coincides with major metabolic changes is the mdx/ApoE KO mouse [32]. ApoE is a lipid transporter predominantly synthesized by the liver, the main organ responsible for lipoprotein metabolism, to coat the surface of several lipid-rich lipoprotein particles, including chylomicron remnants, very-low density lipoproteins (VLDL), intermediate density lipoprotein (IDL), and some high-density lipoproteins (HDL) [63]. Compared to humans, normal mice have very low circulating levels of atherogenic, nonHDL-associated cholesterol. Loss of ApoE, however, causes drastic elevations of circulating chylomicron remnants, VLDL, LDL, total cholesterol and nonHDL cholesterol [64] –the lipoprotein fractions associated with increased cardiovascular risk –due to blunted clearance by the hepatic low-density lipoprotein receptor (LDLR), LDL receptor-related protein (LRP), VLDL receptor, ApoE receptor-2, and gp330, which all require ApoE for proper activity [63, 65].
While ApoE KO mice show little to no muscle phenotype [32, 66], loss of ApoE in mdx4CV mice results in drastic exacerbation of atrophy, wasting and fiber-rich infiltration compared with control mdx mice with low (by human standards) circulating cholesterol levels [32]. The triceps brachii is one of the muscles that show the most exacerbation, along with the gastrocnemius muscle, whereas only a mild exacerbation of mdx-associated cardiac dysfunction was observed (Table 2). Advanced MRI/MRS imaging confirmed that the extreme fatty tissue deposition of mdx/ApoE KO mice resembles that observed clinically in DMD muscle but typically absent in mdx mice [67]. Surprisingly, no exacerbation of diaphragm fibrosis was observed in mdx/ApoE KO mice. Titration of appendicular muscle wasting severity, fibrofatty remodeling, and lethality is possible through dietary cholesterol supplementation. Indeed, a 2% cholesterol-containing chow-based diet further accelerated muscle damage, atrophy, and loss of ambulation compared to a standard 0.2% cholesterol-containing, fat-based diet resulting in complete ambulation dysfunction and humane termination at 8–9mo of age (Fig. 1) [68]. Of note, this early hard endpoint is reminiscent of DMD progression with ambulation dysfunction before cardiorespiratory dysfunction. As treatment of these mice with intestinal cholesterol absorption blocker ezetimibe prevented the severe muscle wasting [68], these results suggest a causal role for circulating cholesterol-rich lipoproteins, i.e. nonHDL, in disease exacerbation in the mdx/ApoE KO mouseand corroborate claims that cholesterol accumulation in the muscles of MD mice contributes to DMD pathogenesis [69, 70]. It must be noted however that ApoE deficiency combined with dietary cholesterol supplementation do not simply cause bulk, random or ubiquitous muscle damage as only certain muscles showed exacerbation; in addition, hypercholesterolemia in dysferlin/ApoE double-null mice resulted in significant rectus femoris fibrofatty remodeling and muscle wasting [71], a muscle not affected in mdx/ApoE KO mice. While the exact origin of this muscle-specific disease exacerbation is unknown, it seems as though nonHDL cholesterol aggravation of mdx muscle wasting likely involves increased fibrosis, inflammation and apoptosis [68]. Combined with the common knowledge that nonHDL particles have profound inflammatory properties, one may raise the possibility that transgenic approaches that heighten muscle damage, rather than blunt muscle repair, may lead to improved humanization of the mdx phenotype.
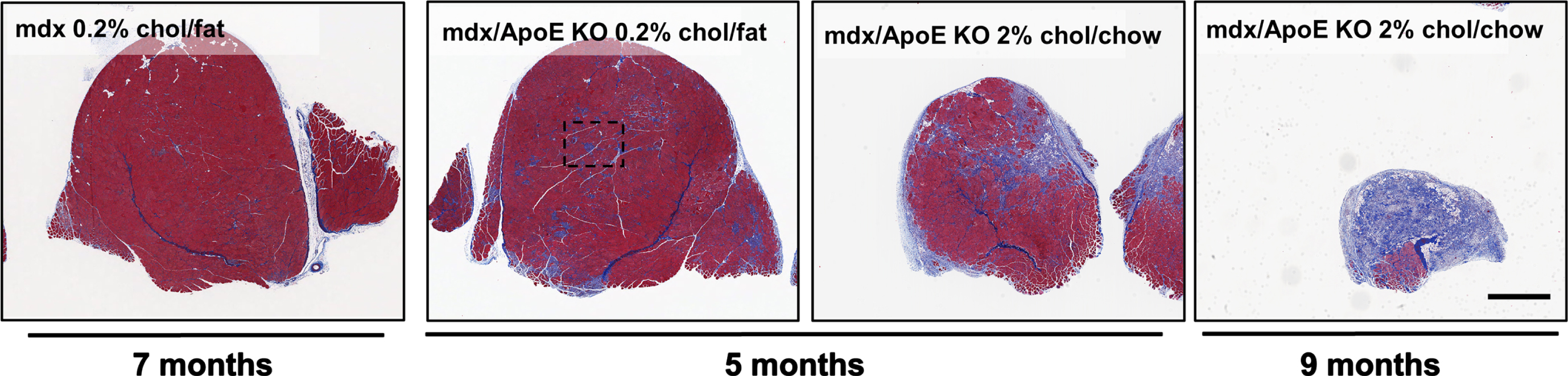
Compared with mdx mice, mdx/ApoE KO mice show evidence of severe wasting, atrophy and fiber-rich infiltration in response to dietary cholesterol supplementation. Mdx mice on an atherogenic 0.2% cholesterol, fat-based diet at 7mo of age show less muscle fibrosis and fibrofatty infiltrates than 5mo mdx/ApoE KO mice on the same diet, whereas cholesterol supplementation with a 2% cholesterol, chow-based diet further exacerbates muscle wasting at both 5 and 9mo of age, the latter resulting in humane termination due to ambulatory dysfunction. Scale bar 1 mm, all images were captured at the same magnification [32, 68].
The relevance of raising circulating nonHDL-associated cholesterol in mdx mice to improve DMD modeling is supported by mounting evidence of major cholesterol abnormalities in MD. Indeed, increases in the cholesterol content of DMD rectus abdominis and gastrocnemius muscle extracts compared with very young control extracts have been reported [72]. We have observed high circulating triglycerides (TGs), nonHDL and LDL-associated cholesterol in 97% of pediatric DMD patients using routine clinical serological testing [33], which supports previous studies that relied on experimental NMR techniques to describe cholesterol-related abnormalities in DMD plasma and muscle samples [73–75]. As unmedicated canine DMD models and female carriers also show circulating lipid abnormalities, in absence of creatine kinase elevation in the case of the latter, high nonHDL, LDL cholesterol and TGs appear to be directly caused by changes in dystrophin expression rather than being secondary to muscle wasting [33]. Dysregulation of miRNAs associated with lipid metabolism and cholesterol synthesis has been documented as a potential cause of muscle wasting in DMD [69]. Hence, DMD is associated with defects in both circulating lipoprotein and intra-myofiber cholesterol homeostasis, as confirmed in a systematic review and meta-analysis [76]. As this metabolic co-morbidity may exacerbate the primary muscle defect [68], this supports the use of transgenic strains with altered lipoprotein metabolism to generate more human-relevant mdx model. Whether non-transgenic approaches that modulate lipoprotein metabolism, such as thermoneutrality or dietary cholesterol supplementation, can also improve mdx modeling of DMD remains to be investigated [77] although non-transgenic rodents are notorious for their low nonHDL levels. Also, as APOE gene mutations are associated with Alzheimer’s disease, [63, 78] reports of Alzheimer’s disease-like symptoms in D2.mdx mice [79] likely provide early evidence of an even more complex link between cholesterol metabolism, cognitive impairment and dystrophinopathies in humans and mice.
To err on the side of caution, it must be noted that despite changing the HDL-rich lipid profile of mdx mice to a more human-like nonHDL-rich profile, loss of ApoE elevates chylomicron remnants and VLDL lipid fractions to levels generally not seen in humans, whereas LDL cholesterol –the predominant nonHDL lipid fraction in humans including DMD patients [33] –is more moderately elevated [32]. Also, although severe ambulation dysfunction of mdx/ApoE KO mice before lethal cardiorespiratory is reminiscent of the disease burden experienced by both untreated and steroid-treated DMD patients, it is not a hard endpoint as what other severe mdx-based mouse models of DMD exhibit. In addition to its role in circulating lipoprotein clearance, intracellular pools of ApoE have been reported and likely play a role in cytoskeleton assembly and stability [80], mitochondrial biogenesis in non-muscle, non-liver cells [81], which may contribute to muscle homeostasis. Since representative and relevant DMD animal models are needed to conduct meaningful treatment and intervention research, it will be important for future studies to understand how mdx/ApoE KO mice respond to established pharmacotherapy, such as prednisone and deflazacort, and to validate the mdx/ApoE KO mouse model for use in drug development.
CONCLUSION AND FUTURE DIRECTIONS
DMD modeling has for a long time relied on the mdx mouse, albeit with well-documented limitations and mixed correlation to human disease severity. As a growing body of evidence suggests that DMD is a complex, multi-system metabolic disease, the possibility of combining multiple transgenic approaches that mimic multiple DMD abnormalities, such as TGF-β signaling and dyslipidemia from a metabolic perspective, but also telomere shortening, a non-metabolic exacerbating factor linked to DMD as shown in the mdx/TERC KO mouse, will likely present new opportunities to better model DMD, study the etiology of this debilitating disease and hopefully refine an already-useful drug testing platform.
CONFLICT OF INTEREST
None.
FUNDING
This work was supported by the Canadian Institutes of Health Research.