
Abstract
Background:
Nemaline myopathy type 6 (NEM6) or KBTBD13-related congenital myopathy is the most prevalent type of nemaline myopathy in the Netherlands and is characterised by mild childhood-onset axial, proximal and distal muscle weakness with prominent neck flexor weakness combined with slowness of movements. The most prevalent variant in the Netherlands is the c.1222C > T p.(Arg408Cys) variant in the KBTBD13 gene, also called the Dutch founder variant.
Objective:
To provide a comprehensive clinical and functional characterisation of three patients to assess the pathogenicity of a newly identified variant in the KBTBD13 gene.
Results:
We present three cases (Patient 1: female, 76 years old; Patient 2: male, 63 years old; and his brother Patient 3: male, 61 years old) with a c.1222C > A p.(Arg408Ser) variant in the KBTBD13 gene. Patient 1 was also included previously in a histopathological study on NEM6. Symptoms of muscle weakness started in childhood and progressed to impaired functional abilities in adulthood. All three patients reported slowness of movements. On examination, they have mild axial, proximal and distal muscle weakness. None of the patients exhibited cardiac abnormalities. Spirometry in two patients showed a restrictive lung pattern. Muscle ultrasound showed symmetrically increased echogenicity indicating fatty replacement and fibrosis in a subset of muscles and histopathological analyses revealed nemaline rods and cores. Slower muscle relaxation kinetics with in vivo functional tests was observed. This was confirmed by in vitro functional tests showing impaired relaxation kinetics in isolated muscle fibres. We found a genealogic link between patient 1, and patient 2 and 3 nine generations earlier.
Conclusions:
The c.1222C > A p.(Arg408Ser) variant in the KBTBD13 gene is a likely pathogenic variant causing NEM6.
INTRODUCTION
Nemaline myopathy type 6 (NEM6) or Kelch repeat and BTB (POZ) domain containing 13 (KBTBD13)-related congenital myopathy is the most prevalent type of nemaline myopathy (NM) in the Netherlands [1]. It is characterised by mild childhood-onset axial, proximal and distal muscle weakness with prominent neck flexor weakness [2–5] (Van Kleef et al., 2024). [6] Importantly, patients with NEM6 may also develop cardiac dysfunction and cardiomyopathy, and respiratory muscle weakness [7, 8]. NEM6 is characterised by two unique features. First, patients exhibit a striking slowness of movements with an inadequately slow motor response to sudden unexpected events and the inability to perform rapidly alternating movements, which is confirmed by slower muscle relaxation kinetics assessed with functional tests [2, 9]. Because of this features, the recently proposed revised NM classification distinguishes NEM6 as a separate form, namely childhood-onset NM with slowness of movements [10]. Second, a particular histological hallmark of NEM6 includes the presence of cores and ring-rods fibres, in addition to nemaline rods, fibre type 1 predominance, prominent nuclear internalisation, nuclear clumps and granulofilamentous protein material [11].
Three autosomal dominant variants in the KBTBD13 gene causative for NEM6 have been found [1, 3, 4, 12]. The c.1222C > T p.(Arg408Cys) is the most prevalent variant in the KBTBD13 gene in the Netherlands and was found in Australian families of Dutch descent, hence it is called the Dutch founder variant [1]. The c.1170G > C p.(Lys390Asn) variant is identified in a Spanish family and a c.199 G>C p.(Gly67Arg) variant in a patient in Italy [1, 4]. A previous study found that KBTBD13 is an actin-binding protein, causing direct structural effects to the actin-based thin filament and modulating muscle kinetics [9].
We here present three patients of two families with a clinical and histopathological NEM6 phenotype with a newly identified c.1222C>A p.(Arg408Ser) variant in the KBTBD13 gene. We evaluated the pathogenicity of this variant by assessing medical history, physical examination, Motor Function Measure (MFM-32), spirometry, muscle ultrasound, muscle biopsy, and in vitro and in vivo functional tests on muscle relaxation kinetics and maximal force. We aim to contribute to the interpretation of the pathogenicity of the newly identified variant in the KBTBD13 gene. This is expected to facilitate the diagnostic process and optimise clinical care for patients with this variant.
MATERIALS AND METHODS
The assessments of the tests were performed at the Radboud university medical centre, Nijmegen, the Netherlands. The majority of the tests of one patient (patient 1) were performed during a cross-sectional study of 42 patients with NM (study protocol number: NL65214.091.18) (Van Kleef et al., 2024). [6] The tests of two patients (patient 2 and 3) were performed in clinical care. The tests were performed in accordance with the Helsinki Declaration of 1975.
We systematically assessed medical history and performed a systematic physical examination including manual muscle testing (Medical Research Council (MRC) (sum) scores) [13, 14], assessment of skeletal abnormalities, and functional clinical tests including the Motor Function Measure 32 (MFM-32) [15]. Spirometry was performed in accordance with the standards of the American Thoracic Society and the European Respiratory Society and compared with reference values [16–18]. Patient 1 and patient 2 underwent a needle muscle biopsy as part of routine clinical diagnostic work-up. The biopsy of patient 1 was included in our histopathological study on NEM6 [11]. The muscle biopsy specimens were processed for routine histological reactions according to standardised methods [11]. The number of rods (including the presence of ring-rods fibres), cores, internalised nuclei, nuclear clumps, fibre splitting and the percentage of type 1 fibres was quantified in accordance with previously described methods [11]. For patient 1, electron microscopic (EM) images were additionally available from our previous study [11]. Muscle thickness and echogenicity (quantitative grey-scale analysis) of a subset of bilateral muscles was assessed by muscle ultrasound in accordance with previously described methods [19–22]. The muscle echogenicity and thickness were standardised by calculating the z-score, corrected for sex, age, length and weight.
To investigate whether sarcomere dysfunction contributes to the muscle weakness and impaired relaxation kinetics, contractile measurements on permeabilized muscle fibres isolated from a muscle biopsy of patient 1 were performed in vitro in a previous study [9]. In short, single muscle fibres were isolated from a quadriceps biopsy of patient 1 and control subjects and permeabilized using 10% Triton-X. The permeabilized muscle fibres were mounted between a force transducer and length motor (Permeabilized Fiber System 1400A, Aurora Scientific Inc., Canada) and activated by exogenous calcium solutions. Absolute maximal force, cross sectional area (CSA), maximal tension (maximal force/CSA), and calcium sensitivity of force generation were measured.
In vivo muscle relaxation rate of the deep finger flexors was assessed through the use of transcranial magnetic stimulation (TMS) over the motor cortex during maximal voluntary contraction (MVC). This induces a brief moment of cortical excitation generating a small increase in force followed by an abrupt halt of descending corticospinal drive to the muscle (i.e. the silent period) causing involuntary relaxation of the targeted muscle. We developed and applied this methodology previously [23–25]. The peak relaxation rate was defined as the steepest negative slope in force during the TMS-induced silent period. This rate was normalised to the force preceding muscle relaxation, resulting in the normalised peak relaxation rate (NpRR). The lower limit of normal (i.e. 5th percentile) for NpRR is 10.1 s–1 and 12.0 s–1 for females and males, respectively [23].
CASE DESCRIPTION
Patient characteristics and medical history
Patient 1 was a 76-year-old woman without comorbidities and a BMI of 24 kg/m2. She was also included previously in our histopathological study on NEM6 [11]. Her initial symptoms started at the age of six years and included running more slowly than her peers, inability to perform pull-ups and effortfully getting up from squatting position. At inclusion, she needed support from her arms when climbing the stairs and getting up from a chair. Getting up from squatting position was impossible without help. NM was diagnosed at the age of 57 years based on the clinical and histopathological phenotype. Her deceased sister also had histologically confirmed NM, but was not genetically tested. Her deceased father had muscle symptoms, but was never investigated.
Patient 2 was a 63-year-old man who was diagnosed with disseminated melanoma (Stage IV), which was stable a few years after immunotherapy. His brother, patient 3, was a 61-year-old man with active treatment for a pituitary adenoma, a possible migrant sensory neuropathy, and obstructive sleep apnoea syndrome. Both patients are morbidly obese, with a BMI of 44 and 42 kg/m2, respectively. Initial symptoms of these patients were similar to those of patient 1, including the slowness of movements. The age of onset was around 10 years. Around the age of 50 years they became unable to run. At inclusion, both patients were unable to lift heavy objects and needed support from their arms when climbing the stairs and getting up from a chair. Getting up from squatting position was impossible without help. They were both diagnosed with NM around the age of 60 years based on the results of muscle biopsy in patient 2 and genetic testing in both patients. Their deceased mother had the same muscle symptoms, according to them, but she never consulted a clinician for this. She had difficulties walking the stairs when patients 2 and 3 were children. Later in life, she also had difficulties walking and could only rise from a chair with support of her arms. She died at the age of 85 years due to heart failure. The c.1222C>A p.(Arg408Ser) variant in the KBTBD13 gene was found post-mortem in her DNA in a previously acquired skin biopsy.
Physical examination
The MRC scores of individual muscles are shown in Figure 1. The MRC sum score (0–60) was 47 in patient 1, 54 in patient 2 and 56 in patient 3. We found generalised muscle weakness with neck flexor weakness. There was no facial muscle weakness, but all patients showed a high arched palate. No other skeletal abnormalities were observed. The functional tests were only performed in patient 1 and 2, as patient 3 could not deliver physical effort due to symptoms of the pituitary adenoma. Both patient 1 and 2 were unable to walk on heels (Video 1) and run. They needed much support of their hands to climb the stairs (Video 2), were unable to get up from a squatting position without help and were not able to jump (Video 3). Patient 2 needed support of his hands to stand up from a chair (Video 4).
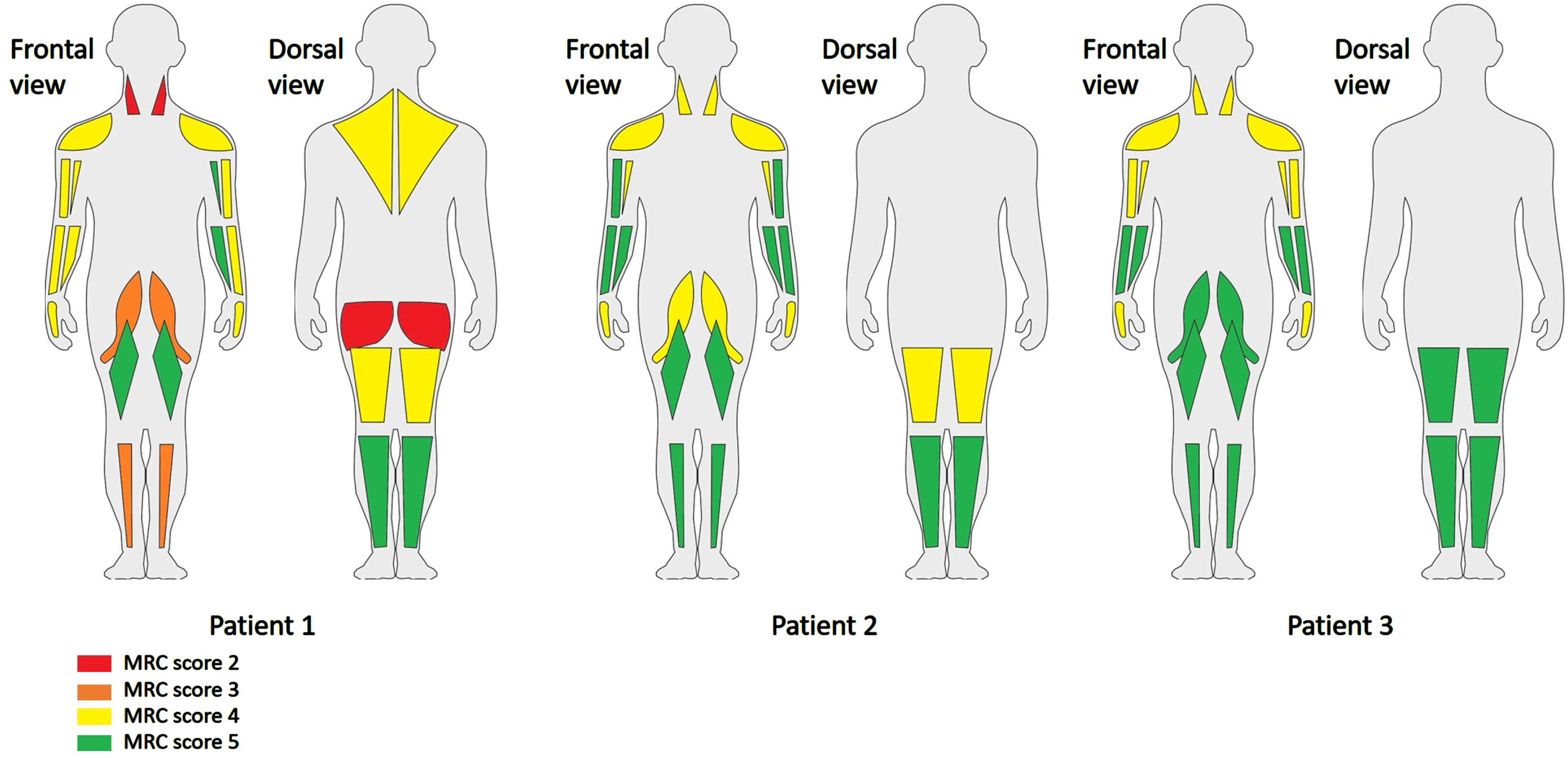
MRC scores of the patients in individual muscles. Legend: As patient 2 and 3 could not turn to their abdomen, the neck and hip extensors were not tested.
Motor function measure
We performed the MFM-32 in patient 1 and patient 2. The total scores were 83% and 55%, respectively. In both cases, domain 1 (standing and transfers) was most severely affected (Patient 1 : 67%, Patient 2 : 26%). Domain 2 (axial and proximal motor function) was affected to a lesser extent (Patient 1 : 92%, Patient 2 : 67%). In contrast, domain 3 (distal motor functions) was (relatively) spared in both patients (Patient 1 : 100%, Patient 2 : 90%).
Spirometry
Spirometry showed an FVC of 75% and 62% of predicted and an FEV1/FVC of 79% and 71% of predicted in patient 1 and 2, respectively. These results indicate a restrictive lung pattern. Spirometry was too strenuous to perform for patient 3.
Genetic findings
The patients harboured the c.1222C>A p.(Arg408Ser) heterozygous variant in the KBTBD13 gene. In patient 2, this was detected by whole exome sequencing with panel analysis. In patient 1 and 3, Sanger sequencing of the KBTBD13 gene was performed, because of the typical NEM6 phenotype and the positive family history, respectively. It was initially classified as a variant of uncertain significance based on the high conservation of the encoded amino acid in orthologue alignments and absence in control populations (gnomAD), but otherwise lack of segregation and functional data. Moreover, the variant is localised in the same codon as the c.1222C > T p.(Arg408Cys) Dutch founder variant. Genealogic research linked patient 1 with patient 2 and 3 nine generations earlier in the 17th century.
Muscle ultrasound
Muscle ultrasound performed in patient 2 and 3 showed an increased echo intensity and decreased muscle thickness in a subset of muscles (Fig. 2). The echogenicity was bilaterally increased (z-score>2.0) in the following muscles in both patients: sternocleidomastoid, flexor carpi radialis, and rectus femoris muscles. The vastus lateralis, the biceps femoris, the medial head of the gastrocnemius, and the tibialis anterior muscles were spared. In patient 2, the deltoid and biceps brachii muscles were affected, while in patient 3 those muscles were spared. Additionally, muscle ultrasound showed generalised muscle atrophy. In particular, the flexor carpi radialis muscle was severely atrophied in both patients. We found no difference in echogenicity and thickness between the left and right sided muscles.
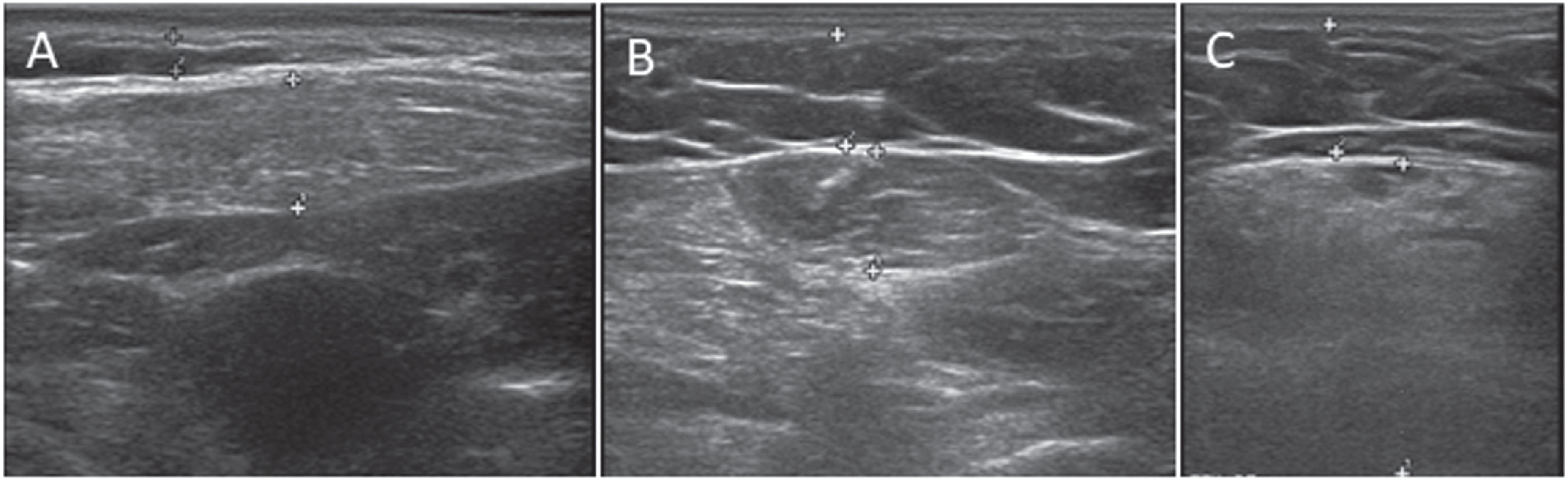
Increased echogenicity on muscle ultrasound. (A) sternocleidomastoid muscle of patient 3. (B) flexor carpi radialis muscle of patient 3. (C) rectus femoris muscle of patient 2.
Muscle biopsy
Patient 1 and patient 2 underwent a muscle biopsy as part of the clinical diagnostic work-up, showing key muscle histology features of NEM6 (Fig. 3) [11]. Both patients showed rods (Patient 1: confirmed on EM, not quantified; Patient 2 : 42%), ring-rods fibres (Patient 1 : 20.95%, Patient 2 : 11.56%), cores (Patient 1 : 71%, Patient 2 : 10%), internalised nuclei (Patient 1 : 50%, Patient 2 : 45%), nuclear clumps (Patient 1 : 13%, Patient 2 : 9%), fibre splitting (Patient 1 : 1,6%, Patient 2 : 0,7%), and fibre type 1 predominance (Patient 1 : 73%, Patient 2 : 56%). There was a large variation in diameter of fibres, as we found atrophy as well as hypertrophy. No clues for alternative histopathological diagnoses were found.
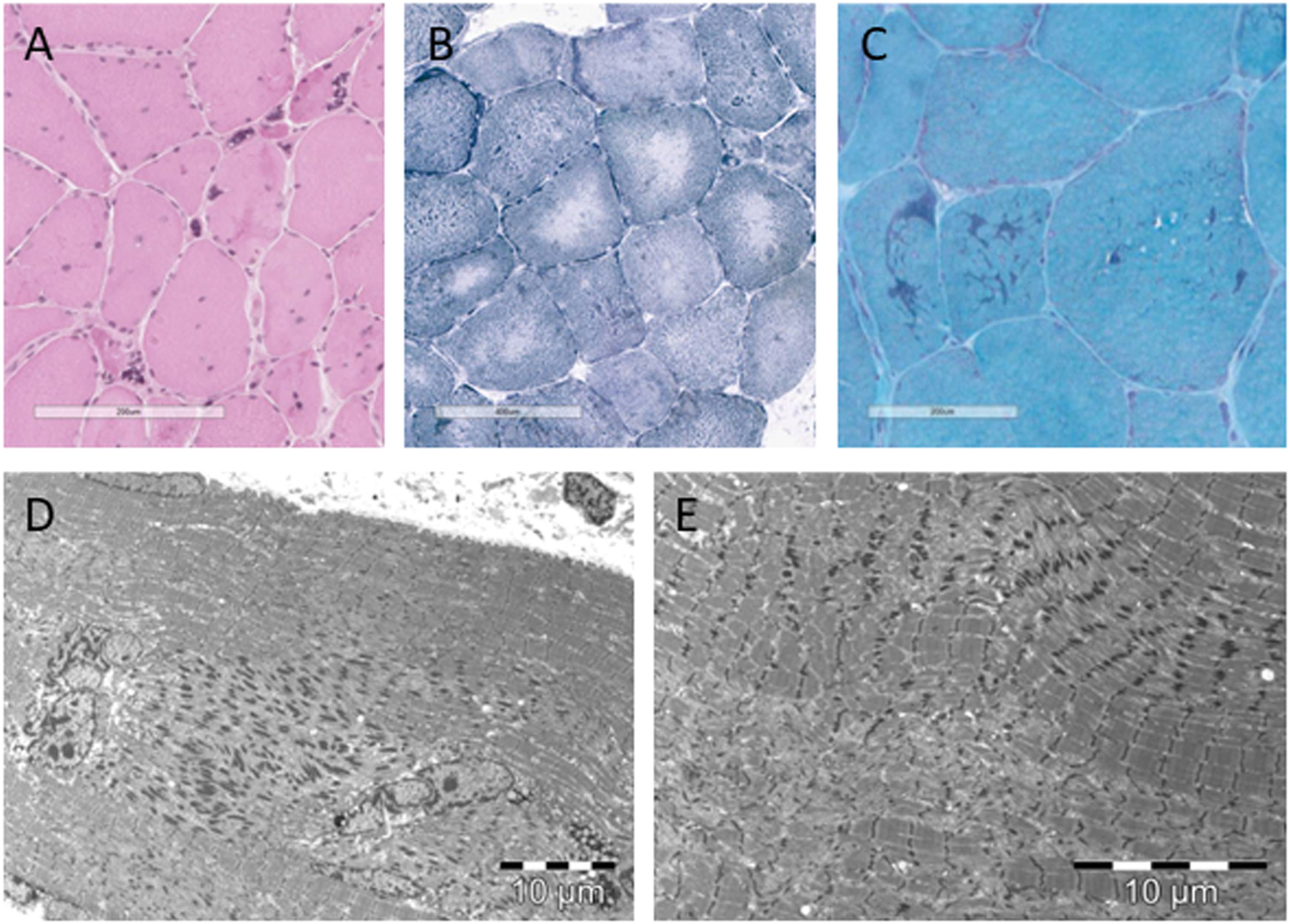
Histopathological characteristics of patient 1. (A) Hematoxylin-eosin staining showing nuclear clumps, internalised nuclei, atrophic and hypertrophic fibres. (B) reduced nicotinamide adenosine dinucleotide tetrazolium reductase (NADH-TR) staining showing cores. (C) modified Gömöri trichrome staining showing rods. (D) electromicroscopic image showing rods and internalised nuclei. (E) electromicroscopic image showing rods and cores.
In vitro and in vivo functional muscle tests
In vitro contractility assays were previously performed on the myofibers of patient 1 [9]. Muscle mechanics on permeabilized muscle fibres revealed that maximal active tension was lower in both slow-and fast-twitch fibres compared with healthy controls (Fig. 4A). Relaxation kinetics were slower in slow-twitch fibres of the patient compared to healthy controls, and in the lower range of healthy controls in fast-twitch fibres (Fig. 4B). Next, single muscle fibres were exposed to incremental calcium solutions to determine the calcium sensitivity of force generation. The force-pCa curve of patient 1 is not affected in slow-twitch fibres, but shifted leftwards in fast-twitch fibres compared to controls (Fig. 4 C). In line with these findings, the pCa50 value was in the range of controls in slow-twitch fibres and above the means of healthy controls in fast-twitch fibres (Fig. 4D), indicating an increased calcium sensitivity of force generation in fast-twitch myofibers of Patient 1.
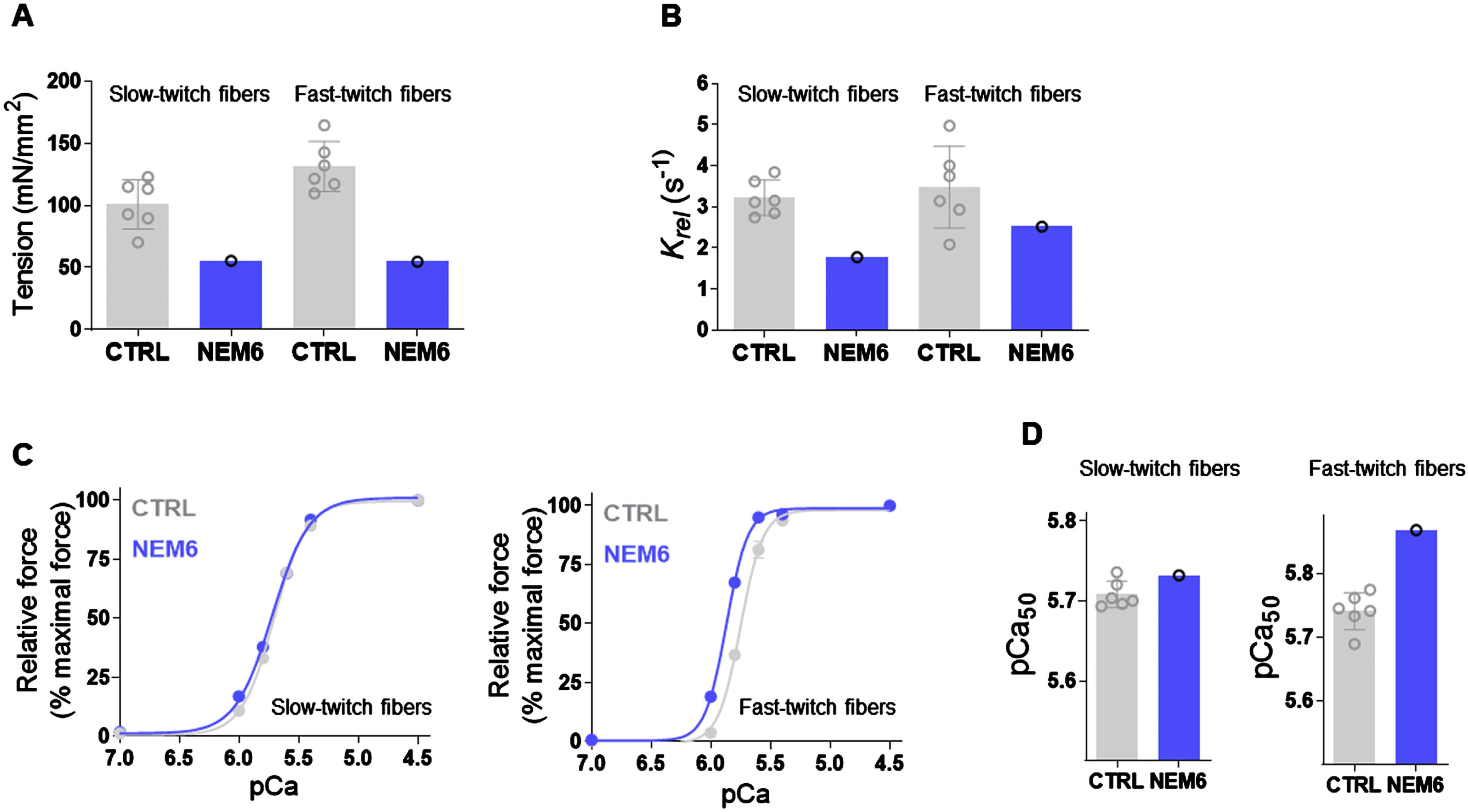
In vitro functional tests showing lower muscle tension, slower muscle relaxation kinetics and increased calcium sensitivity of force generation in patient 1. (A) maximal active tension was lower in both slow- and fast-twitch fibres compared to healthy controls. (B) Relaxation kinetics were slower in slow-twitch fibres of the patient compared to healthy controls, and in the lower range of healthy controls in fast-twitch fibres. (C) The force-pCa curve is not affected in slow-twitch fibres, but shifted leftwards in fast-twitch fibres compared to controls. (D) the pCa50 value was in the range of controls in slow-twitch fibres and above the means of healthy controls in fast-twitch fibres.
In vivo TMS-induced NpRR in patient 1 was 3.8 s–1, which is below the lower limit of normal for females (10.1 s–1). NpRR in patient 2 was 11.5 s–1. This was borderline decreased as the lower limit of normal for males is 12.0 s–1.
DISCUSSION
In this case series, we show that the c.1222C>A p.(Arg408Ser) variant in the KBTBD13 gene is the likely cause of NEM6 in the three studied patients. We found several similarities to patients with NEM6 in the literature with the c.1222C>T p.(Arg408Cys) variant: (1) Childhood-onset of mild muscle weakness and slowness of movements; (2) Generalised muscle weakness with neck flexor weakness; (3) Cores and nemaline rods in muscle biopsy; (4) A restrictive lung pattern tested by spirometry; (5) Increased echogenicity on muscle ultrasound indicative of a muscle disease; (6) In vitro and in vivo functional tests showing lower contractile force of myofibers and slower muscle relaxation kinetics.
Using the American College of Medical Genetics (ACMG) guidelines for variant interpretation [26], the c.1222C>A p.(Arg408Ser) variant in the KBTBD13 gene is considered to be likely pathogenic. It fulfils one criterion of strong evidence (PS) and two criteria of moderate evidence of pathogenicity (PM): (1,PS3) We used in vitro and in vivo functional tests supportive of a damaging effect of the gene product; (2,PM2) The variant has not been found in a control group of patients in the Genome Aggregation Database; (3,PM5) This is a novel missense change at an amino acid residue where a different missense change determined to be pathogenic has been seen before, i.e. the c.1222C>T p.(Arg408Cys) variant. As additional supportive evidence (PP3) we identified that multiple lines of computational evidence support a deleterious effect on the gene as a high conservation of the encoded amino acid is found.
In accordance with patients with NEM6 from the literature and our Dutch NEM6 cohort [2–5], we found childhood-onset of mild axial, proximal and distal muscle weakness with neck flexor weakness and slowness of movements. In more detail, these patients showed similar childhood-onset difficulties in performing sport-related activities, which progressed into functional abnormalities in adulthood, including climbing stairs and getting up from a chair. Moreover, the MFM-32 showed that domain 1 (standing and transfers) was most severely affected, while domain 3 (distal muscle weakness) was relatively spared.
Muscle ultrasound showed increased echogenicity indicating fatty replacement and/or fibrosis in a subset of muscles, confirming the presence of a neuromuscular disease. No previous studies on muscle ultrasound in patients with NEM6 have been performed so far. Muscle imaging studies (muscle MRI or CT) showed predominant involvement of thigh muscles, the lateral and medial gastrocnemius muscles, peroneal group and soleus muscle with less prominent changes in the anterior tibialis muscle [4]. In our study using muscle ultrasound, the rectus femoris muscle was indeed affected, while the tibialis anterior muscles were spared. Interestingly, the medial head of the gastrocnemius, the vastus lateralis and the biceps femoris muscles were not affected in the two patients included in this case series. This discrepancy in muscle involvement can be caused by interpatient variations and/or by the differences inherent to muscle ultrasound and MRI [22, 27].
The histopathological findings of the presented patients are in accordance with those of NEM6 caused by the c.1222C > T p.(Arg408Cys) variant [4, 11]. These histopathological features include the presence of rods, cores, internalised nuclei, nuclear clumps, myofiber splitting, type 1 fibres predominance, and a large variation in diameter of fibres, in the absence of histological clues for other myopathies. One of the patients included in our study on the extensive description of the histopathological features of NEM6 was also included in the current study [11]. Rods are a typical hallmark for NM in general [28].
In vitro tests showed lower muscle tension, slower muscle relaxation kinetics and increased calcium sensitivity of force generation. This lower muscle tension can represent the lower muscle force, while the slower muscle relaxation kinetics combined with the increased calcium sensitivity may contribute to the typical slowness of movements that is experienced by the patients. The findings of our in vitro tests are thus in line with our in vivo findings, and support the pathogenicity of the found variant in the KBTBD13 gene.
The genealogic link between patient 1, and patient 2 and 3 nine generations earlier, strongly suggest that there are more people with the c.1222C > A p.(Arg408Ser) variant in the KBTBD13 gene and a NEM6 phenotype. Moreover, the muscle symptoms in the family members also suggest this. Very likely, the relatively mild phenotype leads to a largely unrecognised neuromuscular disorder. This is in line with the late age at diagnosis of the patients in this case series. Since the parents were not alive anymore, we could not assess the penetrance. Based on family history, the penetrance is at least very variable. In parallel, in families with the c.1222C > T p.(Arg408Cys) variant the disease was identified merely two decades ago [2]. Since then NEM6 has become more known among health-care providers and families, leading to an earlier recognition. This shift will probably need to take place in families with the newly identified variant.
Previously, additional variants in the KBTBD13 gene were identified as causative for NEM6, namely the c.742C > A p.(Arg248Ser) variant and the c.742C > T p.(Arg248Cys) variant [1, 11]. However, the prevalence of these variants in control populations in the gnomAD database is too frequent to be considered causative for NEM6. Therefore, these variants are currently identified as ‘likely benign’. Thus, the c.1222C > A p.(Arg408Ser) variant reported in our study, is currently one of the four variants to be likely causative of NEM6. The other variants are c.1222C > T p.(Arg408Cys), c.1170G > C p.(Lys390Asn), and c.199 G>C p.(Gly67Arg) as mentioned earlier [1, 3, 4, 12].
There are several limitations of this case series. First, only three patients were identified with this KBTBD13 gene variant and we were not able to perform the same tests in all patients. This is a challenge for the confirmation of pathogenicity. However, the many similarities with patients with diagnosed NEM6 in combination with the fulfilled ACMG criteria, enabled us to consider the variant to be likely pathogenic. Second, the elderly age of patient 1 and the presence of morbid obesity in patient 2 en 3 probably affected the experienced symptoms and physical tests. However, the muscle biopsy and ultrasound results, and the TMS-induced muscle relaxation are not affected by or are corrected for these factors. Moreover, the patients experienced muscle symptoms since childhood and the physical tests were more severely affected than expected based on the age or BMI.
In conclusion, we show that there are many similarities between patients with the c.1222C > A p.(Arg408Ser) heterozygous variant in the KBTBD13 gene and patients with NEM6 with the c.1222C > T (p.Arg408Cys) variant. According to the ACMG criteria the variant is likely pathogenic. Hence, the variant can be identified as a new likely causative variant of NEM6. This is an addition to the limited number of variants identified thus far [1, 4]. This case series is expected to result in the identification of more patients with this variant. This is important for diagnosing patients, providing medical care for cardiac and respiratory manifestations that can occur in NEM6, and informing families about the recurrence risk/genetic aspects [7, 8].
Supplemental Material
Supplementary Video 1
Supplemental Material
Supplementary Video 2
Supplemental Material
Supplementary Video 3
Supplemental Material
Supplementary Video 4
Footnotes
ACKNOWLEDGMENTS
The patients included in this case series are acknowledged for their participation. Sultan Bastu, research engineer, is acknowledged for additional analyses of the muscle biopsies.
Several authors of this publication are members of the Radboudumc Center of Expertise for neuromuscular disorders (Radboud-NMD), Netherlands Neuromuscular Center (NL-NMD) and the European Reference Network for rare neuromuscular diseases (EURO-NMD).
Funding
This work was financially supported by the Princess Beatrix Fund (Grant number W.OR17-08) and A Foundation Building Strength.
Conflict of interest
The authors have no conflict of interest to report.
Ethics approval
We hereby confirm that the present study conforms to the ethical standards and guidelines of the journal and is in accord with the Helsinki Declaration of 1975. Patients granted permission for publication of their medical data.
Data availability statement
The data that support the findings of this study are available within the article, and from the corresponding author, upon reasonable request.