
Abstract
Facioscapulohumeral muscular dystrophy (FSHD) is an exclusively human neuromuscular disease. In the last decades the cause of FSHD was identified: the loss of epigenetic repression of the D4Z4 repeat on chromosome 4q35 resulting in inappropriate transcription of DUX4. This is a consequence of a reduction of the array below 11 units (FSHD1) or of a mutation in methylating enzymes (FSHD2). Both require the presence of a 4qA allele and a specific centromeric SSLP haplotype. Muscles become involved in a rostro-caudally order with an extremely variable progression rate. Mild disease and non-penetrance in families with affected individuals is common. Furthermore, 2% of the Caucasian population carries the pathological haplotype without clinical features of FSHD.
In order to explain the various features of FSHD we applied Ockham’s Razor to all possible scenarios and removed unnecessary complexities. We postulate that early in embryogenesis a few cells escape epigenetic silencing of the D4Z4 repeat. Their number is assumed to be roughly inversely related to the residual D4Z4 repeat size. By asymmetric cell division, they produce a rostro-caudal and medio-lateral decreasing gradient of weakly D4Z4-repressed mesenchymal stem cells. The gradient tapers towards an end as each cell-division allows renewed epigenetic silencing. Over time, this spatial gradient translates into a temporal gradient based on a decreasing number of weakly silenced stem cells. These cells contribute to a mildly abnormal myofibrillar structure of the fetal muscles. They also form a downward tapering gradient of epigenetically weakly repressed satellite cells. When activated by mechanical trauma, these satellite cells de-differentiate and express DUX4. When fused to myofibrils they contribute to muscle cell death in various ways. Over time and dependent on how far the gradient reaches the FSHD phenotype becomes progressively manifest. We thus hypothesize FSHD to be a myodevelopmental disease with a lifelong attempt to restore DUX4 repression.
BACKGROUND
It took 20 years from the discovery of the gene location on chromosome 4q35 to the publication of a unifying genetic model explaining the pathophysiology of FSHD (Fig. 1) [1, 2]. This model shows that epigenetic de-repression of the D4Z4 repeat and its product DUX4 is the cause of FSHD.
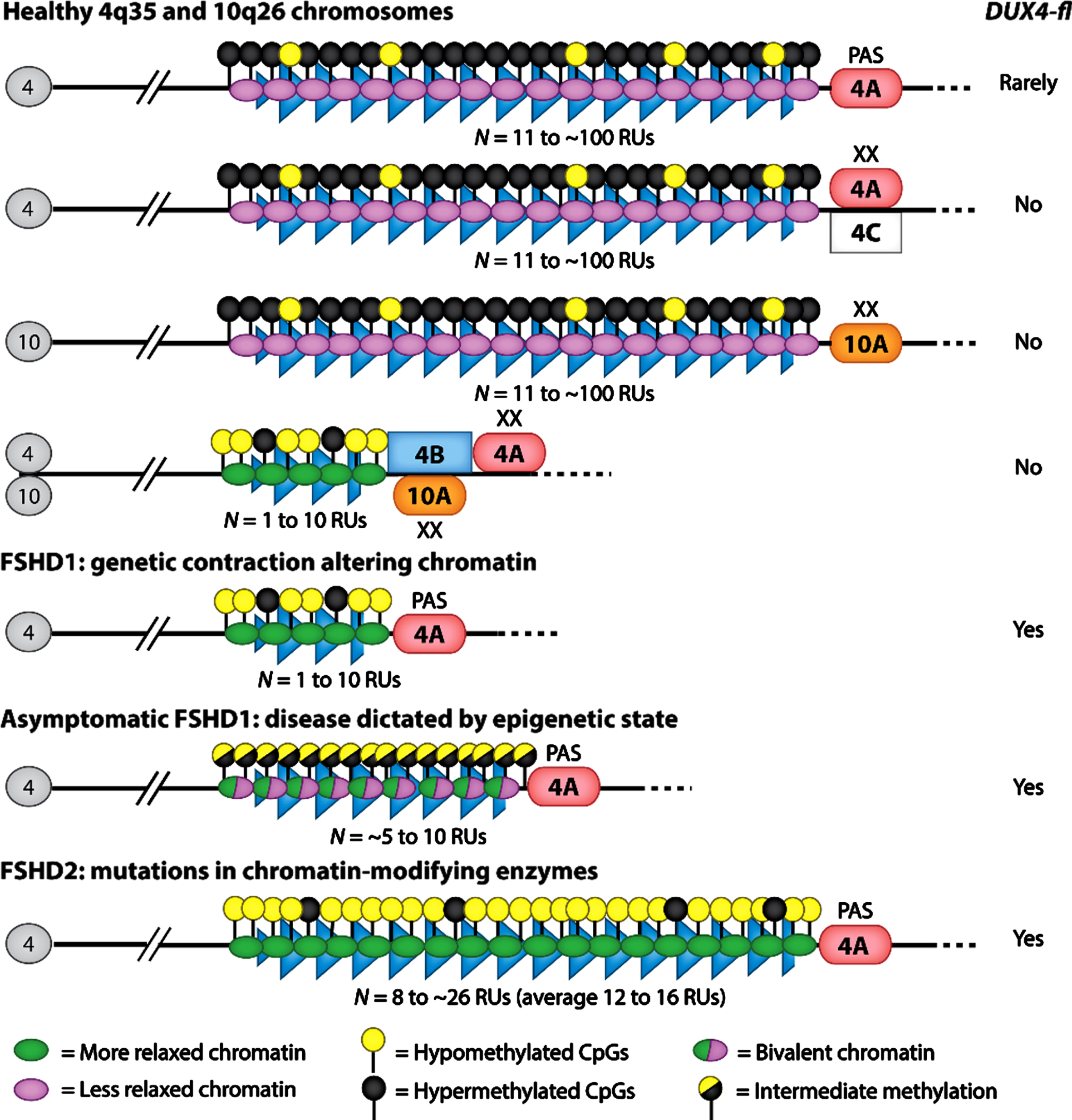
The genotype of FSHD is extremely complicated. The genetics and epigenetics of facioscapulohumeral muscular dystrophy (FSHD), depicting the FSHD1 and FSHD2 alleles compared with a spectrum of healthy alleles. The D4Z4 macrosatellite arrays at standard chromosomes 4q35 and 10q26 contain from 11 to approximately 100 repeat units (RUs) (blue triangles). In FSHD1 patients, the array is contracted to 1–10 RUs on one 4q35 allele. FSHD2 patients display slightly shortened arrays (8 to approximately 26 RUs) mostly within the typically healthy range. The telomeric region distal to the array exists as two prominent alleles: 4qA and 4qB. Rare chromosomes lacking A or B are referred to as 4qC. In healthy individuals, the array is marked by DNA hypermethylation and chromatin compaction, indicating a state of transcriptional repression. In both FSHD1 and FSHD2, the array displays DNA hypomethylation and chromatin relaxation, indicating a state that is more permissive for gene expression. Asymptomatic individuals display an epigenetic profile that is intermediate between unaffected and affected. The DUX4 gene is encoded within each RU of the D4Z4 array. Both forms of FSHD require disease-permissive haplotypes of 4qA (containing a polyadenylation signal for DUX4) and at least 1 RU. Nonpermissive haplotypes on either 4qA or 10qA do not result in FSHD. The pathogenic full-length DUX4 transcript (DUX4-fl) is expressed in both forms of FSHD and occasionally in asymptomatic subjects, whereas expression is very rare or undetectable in healthy individuals. Additional abbreviation: PAS, polyadenylation signal [2]. Reprinted from Annu Rev Genomics Hum Genet., Himeda CL, Jones PL., The Genetics and Epigenetics of Facioscapulohumeral Muscular Dystrophy, 2019;20:265-291, with permission of Annual Reviews (US). The publication is available through http://dx.doi.org/10.1146/annurev-genom-083118-014933.
There are several reasons why this understanding took so long. First, the D4Z4 repeat has complex interactions with an almost identical repeat on chromosome 10q26. Furthermore, the genetic preconditions needed for stable DUX4 transcripts (4qA haplotype and hypomethylation of the repeat) still had to be discovered. Subsequently, some epigenetic modifiers of the repeat were identified, and the specifics of the haplotype and the polyadenylation signal (PAS) were sorted out in more complex models [2–5]. However, the current model [1] is still not sufficiently detailed to explain how the repeat is repressed or de-repressed. A second reason for the long road to the unifying model is the very sporadic presence of DUX4 in muscle biopsies. Its production must be inferred by the demonstration of activation of downstream factors: RNA and protein expression studies in myoblast cultures and muscle biopsies have identified a large number of dysregulated pathways, many of which by themselves can lead to muscle cell death [6]. A third complicating factor is the fact that FSHD is a strictly human disease. Hence, a natural animal model does not exist. The different animal constructs all have their limitations and cannot faithfully produce the FSHD phenotype [7]. A fourth factor is the observation that the pathological 4qA haplotype is found (apparently repressed) in 2% of the normal Caucasian population [8], and (also completely repressed) in 10–30% of the non-penetrant sibs with the pathological haplotype [9, 10]. Moreover, it is also assumed to be repressed in the lower limb muscles of the 30% of manifesting carriers who never develop lower limb involvement [11].
The phenotypic variability is often explained as the consequence of different upstream factors involved in D4Z4 repression plus the polymorphisms and metabolic variances of the downstream targets [6]. However, it is unlikely that these variables act stochastically in the void or that they replay a part of the developmental body plan. More likely, they act upon a template, a scaffold, in a similar way as the changes in the plasmalemma act as a scaffold for the unfolding pathology in Duchenne muscular dystrophy [12]. These considerations inspired us to understand FSHD pathogenesis from the opposite side of the muscle demise, and see FSHD as the result of the failure of the lifelong attempt to restore the repression of D4Z4 in all possible tissues and cells.
HYPOTHESIS: FSHD AS A MYODEVELOPMENTAL DISEASE
We will summarize the lessons that can be learnt from FSHD genotype, phenotype, muscle morphology and molecular biology. We have applied Ockham’s Razor to all possible scenarios and removed unnecessary complexities. This resulted in a hypothesis on FSHD pathology that best fits these data: FSHD pathology reflects the ultimate failure of a lifelong attempt to keep the repeat silenced. The template that is laid down early in embryogenesis determines the phenotypic variability, i.e. the possible extent of expression of the FSHD phenotype. Epigenetically weakly DUX4-repressed muscle stem-cells contribute to a restricted, mild congenital myopathy in the trunk and limbs. This myopathy diminishes in severity in a rostro-caudal gradient, which extent is roughly inversely related to the size of the D4Z4 repeat. It contains a subset of similarly weakly repressed satellite-cells. We propose that these cells, when activated later in life by mechanical trauma induced by physical activity, de-differentiate and briefly express DUX4, inducing the FSHD specific pathological process.
The genotype of FSHD is extremely complicated (Fig. 1) and its epigenetic regulation is still not fully understood. We will discuss some blanks in the knowledge of repression and de-repression of the repeat.
FSHD is clinically an autosomal dominant progressive muscle disease that comes in two genetic subtypes both caused by epigenetic de-repression of a 3.3 kb repeat array on chromosome 4q35 [4, 7]. In controls the array contains 8–100 units. In FSHD1 (95% of all cases), the array consists of 1–10 units and is hypomethylated. Each repeat contains an open reading frame of a double homeobox sequence. The distal repeat can be stably transcribed only in the presence of a telomeric 4qA sequence and a permissive more centromeric single sequence length polymorphism (SSLP) variant. The 4qA allele contains the pLAM sequence with a polyadenylation signal (PAS) and a 68kb ß-satellite array. In contrast, 4qB alleles have no functional PAS and are thus not pathogenic. In the normal population both allelic forms are present in equal numbers [1, 4].
An almost identical repeat array resides on chromosome 10q26. In the absence of a functional PAS, this does not lead to a stable transcription. The 4q35 and 10q26 repeats can be distinguished based on restriction enzyme characteristics. The repeats are reported to be exchangeable. In the Caucasian population roughly 5% of the 4q arrays harbor 10q repeats and vice versa. An excess of 4q type repeats on chromosome 10 was suggested to be a premutation condition and the mutation to be caused by inappropriate CTCF- and cohesion binding followed by loop-excision [13].
In FSHD1 the mechanism of mutation was reported to be a mitotic intra-chromatid repeat exchange in germ cells or early in embryogenesis, leading to a reduction of repeats in the D4Z4 array on chromosome 4 [14]. When this mitotic mutation mechanism happens early in the zygote it is responsible for mosaic gene carriers with variable signs and symptoms. The mosaicism explains the unusual combinations of a minimally affected parent with a severely affected child [13, 15]. The reduction of the repeat leads to its hypomethylation in a manner not precisely known. Furthermore, the extent of hypomethylation is not linearly related to the severity of the disease. Hence, hypomethylation of the repeat is conditional but not causal for the disease.
FSHD2 (5% of all cases) is a digenic condition caused by heterozygous mutations in the methylating enzymes SMCD1 (structural maintenance of chromosomes flexible hinge domain containing 1) or DNMT3B (DNA methyltransferase 3 beta), or by homozygous deletions in LRIF1 (ligand dependent nuclear receptor interacting factor 1), an SMCHD1-protein interactor. FSHD2 only becomes manifest in the presence of 8 to 26 D4Z4 repeat units on a 4qA type chromosome. In FSHD2 hypomethylation is present on both chromosomes 4 and 10. Finally, mutations in the FSHD2 genes can act as modifiers of FSHD1 [2, 5].
The transcription of the last D4Z4 repeat-unit is complex and encodes for two proteins. DUX4-fl (full length) is a protein of 424 amino-acids that contains the double homeobox and the C-terminal domain, required for most metabolic activities. DUX4-s (short) is a protein of 159 amino-acids that lacks the C-terminal domain and is not pathogenic [3]. DUX4 has a function in Zygote Genome Activation (ZGA), using a different PAS in exon 7 available to both A and B type alleles [16]. During the early steps of embryogenesis, DUX4 is involved in the regulation of genes important for pre- and post-implantation development. Subsequently, DUX4 expression is silenced in most adult tissues, with the exception of testis and thymus [4, 5], where it performs largely unknown functions [17].
The epigenetic repression of D4Z4 is a regulated interactive process of a large number of factors involved in histone modifications and DNA methylation. In the de-repression machinery factors of the Trithorax- and Chromatin-Remodeling-Complexes are at work, possibly in conjunction with long non-coding RNA-sequences (lncRNA). Several of these factors have been identified. While a model of the final state of de-repression has been proposed, the trigger and the complex processes from repression to de-repression and back need to be worked out [2, 18]. Several studies have reported double negative and positive feedback loops enabling augmentation of DUX4 expression [5, 19]. However, this research is complicated by the fact that demonstrating DUX4 presence in biopsies and cell cultures by immunohistochemistry is extremely difficult. Transcription is usually inferred by demonstrating expression of downstream target genes.
Family studies suggest that the gene for FSHD is not lethal and does not affect fertility. In general, the disease is characterized by a normal life expectancy and a high spontaneous mutation rate: a population study reported 9% of living gene carriers to be the first patient in their families [11, 20]. Large families with FSHD have become rare. In a population study the average family had five to seven affected living members [21]. Affected sibs abstaining from offspring [11], non-penetrant cases and more recently prenatal and pre-implantation testing are probably better explanations for the disappearance of the disease from these families [10, 22].
From these data we conclude that the vast amount of genetic research in FSHD has unraveled the genetic condition required for FSHD. Yet the occurrence of non-penetrant sibs argue that the genetic condition in itself is not enough for disease expression. More than the shared genetic variations in the downstream targets of DUX4, loss of epigenetic control of the repeat appears responsible for the variable severity of the phenotype. This notion is strengthened by observations of the muscle phenotype (next paragraph). These observations are compatible with the hypothesis that the epigenetic control mechanism does is utmost best to keep DUX4 repressed in muscle. When expressed, the control mechanisms try to keep expression at a minimum. In most cases, this control mechanism succeeds and FSHD does not manifest In some individuals, it fails and symptoms will progressively manifest.
Lessons learnt from the phenotype: Insights from the pattern of muscle weakness
In this paragraph we will discuss the clinical picture of FSHD and stress how little we know of the earliest phases and minimal expression of the disease. Such a focus is relevant to our hypothesis of secondary silencing of DUX4.
Most clinical studies of FSHD are from tertiary referral centers with a selection bias for more advanced disease. They focus on the genetics, the progressive nature of the condition and highlight unusual aspects and associations. As a result, mild disease manifestations generally receive less attention in the medical literature. Furthermore, long-term follow up studies are not available. The course of the disease over time from minimal (only facial weakness), to mild (facial and shoulder girdle muscle involvement), moderate (lower limb involvement) and severe FSHD (wheelchair use) is reconstructed largely based on reported symptoms. This is reflected by the severity scores used in the clinic and in research [9, 24]. We will here describe the steps in this descending course of muscle involvement.
Facial weakness is considered to be an early feature of FSHD. It is often asymmetrical without a side preference [11]. The asymmetry may leave one side with normal or almost normal function which is relevant considering its pathogenesis [25]. Its severity is roughly inversely related to the number of residual repeats and less prevalent in patients with small deletions [26, 27]. Personal observations of patients over longer periods of time have not shown convincing progression of the facial weakness (Padberg, van Engelen, Voermans personal communications). Faces change over time as part of a normal ageing process, but not any elderly patient has been reported to have reached complete facial paralysis later in life, in contrast to axial and extremity muscles. Our ongoing natural history study will provide information on the long term follow-up of facial weakness. Furthermore, we are currently studying facial muscle involvement by qualitative muscle ultrasound. Unfortunately, data on facial muscle morphology are lacking.
Severe facial weakness to the extent of misdiagnosis as Moebius syndrome has been described in early onset, severe FSHD. Childhood photographs of these patients often show an open mouth and occasionally small tongues [28]. However, only few studies on masseter and tongue strength are available [29]. The general impression is that masseter and tongue involvement in early onset severe FSHD is only mildly abnormal, probably congenital and not progressive.
The second earliest sign of the disease is shoulder girdle muscle weakness, often in an asymmetric pattern with, according to many reports, a weaker right (dominant, more active) side [30]. An asymmetric position of the scapula might be visible five to ten years before shoulder and arm dysfunction is recognized. This is likely related to the limited demands of forceful activities of these muscles in daily life and the slowly progressive nature of the disease. This of course contributes to the difficulties pinpointing the onset of the disease.
Overall, most clinical papers testify to the great variability in age of onset and rate of progression of the disease. Most studies report onset in adolescence which is the end of the second phase of muscle growth in humans [31]. If progression to lower limb involvement occurs it takes on average 15 to 20 years with very wide ranges. Of the patients with lower limbs weakness, approximately 20% will become wheelchair dependent later in life [11].
The phenotypic spectrum of gene carriers with the same D4Z4 deletion can best be seen in large and completely examined families. They show that one-third of the gene-carriers older than 50 years does not have recognizable lower limb involvement. Furthermore, 30% of all gene-carriers report no symptoms but might have recognizable signs of the disease on examination. (Fig. 2) [9–11]. We deduced likelihood curves for reporting symptoms and showing signs of FSHD: Fig. 3. This clearly shows that the likelihood of manifestation is age- and deletion-dependent [10].
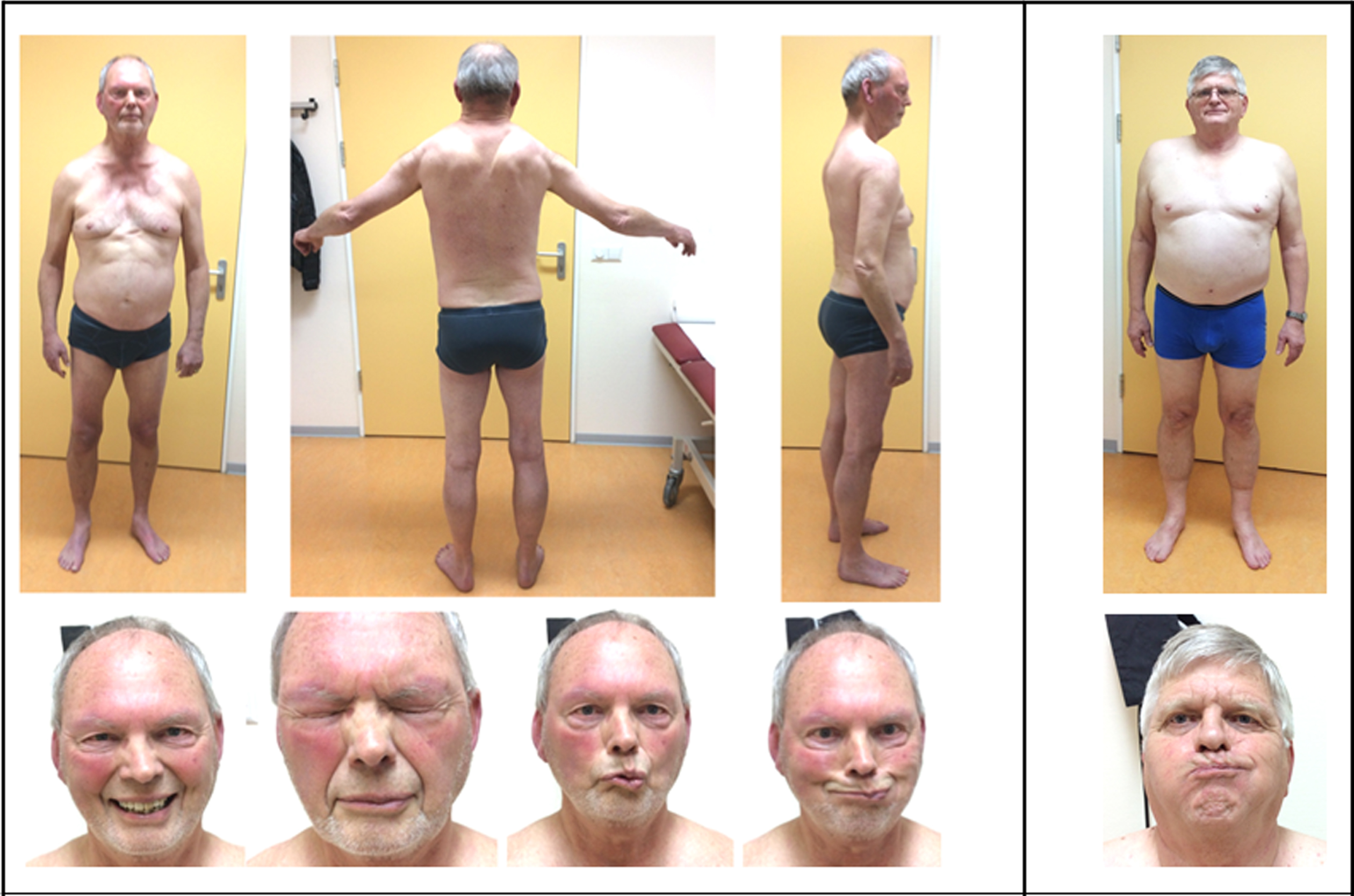
Clinical features of FSHD. Left box: 70-year FSHD patient (genetically confirmed FSHD: D4Z4 repeat of 8 units). He had a clinical severity score (CSS) of 7 at the age of 70. He has had difficulties with gymnastics in school, motor limitations in military service, and was clinically diagnosed with FSHD in his 30 s. Muscle weakness has progressed in a rostro-caudal gradient. Right box: the brother was investigated at our centre because of seemingly incorrect results of segregation analysis. He had worked in construction until the age of 65 and had never reported muscle symptoms. On physical examination, he had facial asymmetry but no other signs of FSHD1 (CSS score 1). This family illustrated the intrafamilial variation in disease severity. Permission granted by the patients.
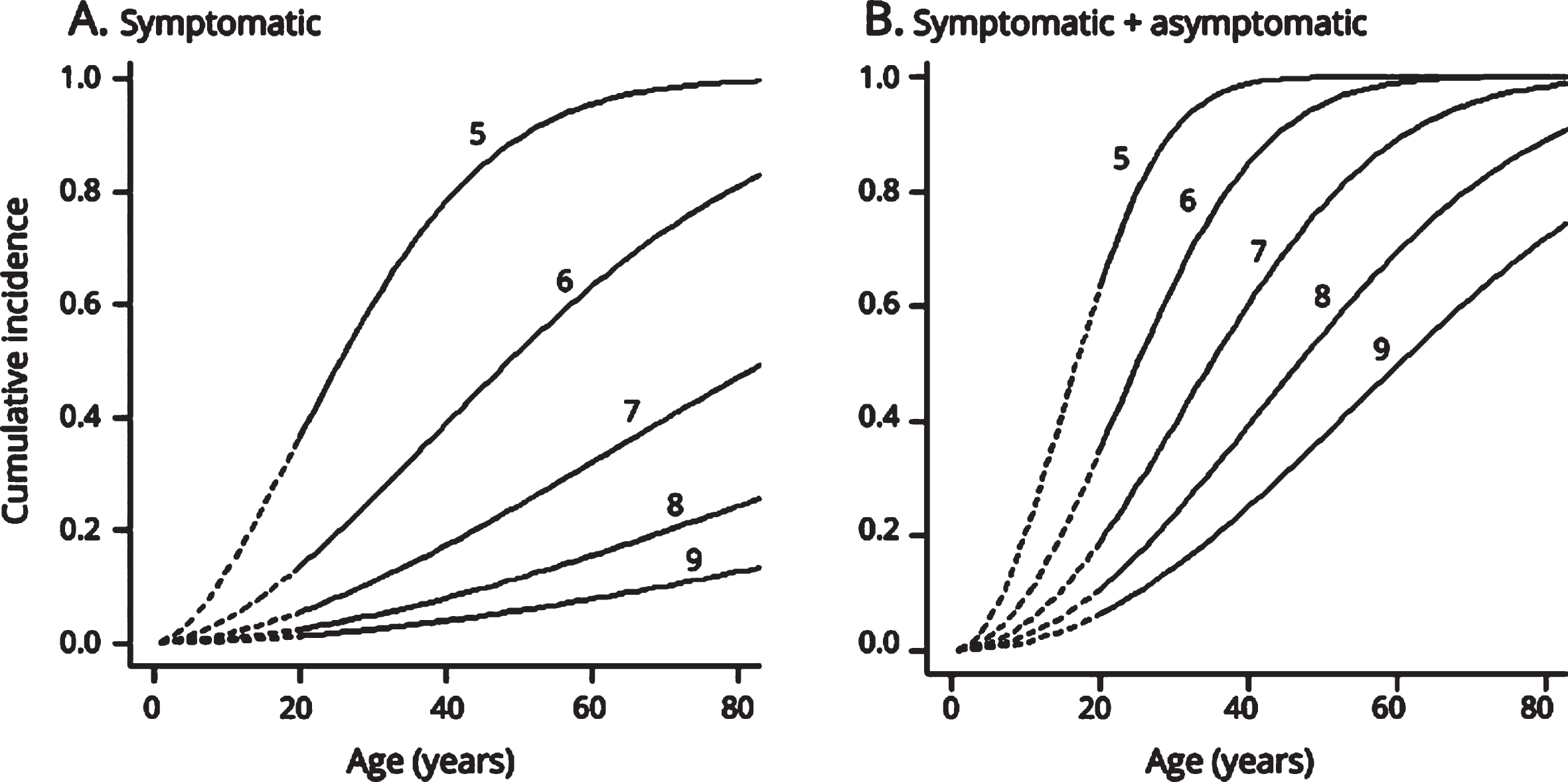
The likelihood of clinical manifestations is age- and repeat contraction-dependent. Maximum likelihood curves of the penetrance. Maximum likelihood estimates of the penetrance of symptomatic (A) and symptomatic plus asymptomatic facioscapulohumeral muscular dystrophy (FSHD) (B) for 5, 6, 7, 8, and 9 D4Z4 units (from up to down) for age. This represents the likelihood of reported symptoms by the patient at a certain age (A: symptomatic mutation carriership) and of reported symptoms by the patients or observed signs by the neurologist at a certain age (B: symptomatic and asymptomatic mutation carriership). Both penetrance likelihoods were modeled as Cox regression models with a Weibull baseline distribution and the logarithm of the number of D4Z4 units as a covariate. (A) Carriers with 8 repeats have a 20% (0.198) chance of being symptomatic at age 70. (B) Carriers of repeat size of 8 units have only a 24% (0.236) chance of being detected by clinical examination at age 30. These likelihood estimates are helpful in counseling; however, the number of patients on which the estimates are based calls for cautiousness. Reprinted from Neurology, Wohlgemuth M, Lemmers RJ, Jonker M, van der Kooi E, Horlings CG, van Engelen BG, van der Maarel SM, Padberg GW, Voermans NC. A family-based study into penetrance in facioscapulohumeral muscular dystrophy type 1, 2018;91:e444-e454. The publication is available through http://dx.doi.org/10.1212/WNL.0000000000005915.
The clinical observations that FSHD does not manifest at all or does not spread to the lower extremities in a substantial number of gene-carriers suggest a successful repression of the repeat despite its contracted state. Even more convincing arguments for the primate of epigenetic regulation in this disease comes from identical twin studies showing different phenotypes in twin pairs [32]. This observation raises the questions what muscle morphology shows in non-manifesting carriers, which we will discuss in the next paragraph.
Abnormalities of muscle morphology were always assumed to be part of an ongoing pathological process reflected by the clinical muscle condition. We would like to consider that minimal abnormalities such as small atrophic fibers might reflect a congenital condition of the disease.
Muscle biopsies were initially performed in search for a characteristic feature for FSHD or to exclude differential diagnosis (in the pre-genetic era). No specific morphological signature was detected. Furthermore, 30% of the muscle biopsies in FSHD did not reveal abnormalities [11]. The biopsies were usual open biopsies taken from standard sites (deltoid muscle, vastus lateralis muscle). With our current hypothesis, this raises the question whether these biopsies were taken from muscles that would never have shown pathology. Consequently, FSHD was considered to be a multi-focal myopathy expressing morphological abnormalities in clinically affected muscles only. Based on the prevalence of observed abnormalities, variation in fiber size and increased extracellular matrix deposition (fibrosis) were considered the earliest microscopic changes in FSHD [11].
Recent studies of muscle morphology and pathophysiology are driven by MRI-guided muscle biopsies. Biopsies of clinically and radiologically affected muscles often show fatty replacement and necrotic fibers [33]. The most characteristic features are likely the perivascular and interstitial cellular infiltrates. The composition of these infiltrates are considered to be specific for FSHD [33–36]. Lobulated fibers, at one time thought to be a hallmark of the disease, were later found to occur in other myopathies as well [11]. These fibers showed subsarcolemmal accumulation of abnormal mitochondria on electron microscopy. Another study reported subsarcolemmal non-specific changes [37]. Cytoplasmic calcium accumulation was seen in a few cells in FSHD [38]. A recent study demonstrated hypovascularity in muscle biopsies [33, 35]. A number of these observations need corroborating studies. Finally, DUX4 positivity is rarely found or absent in most biopsies, but apoptotic features have been reported [4, 33].
It is hard to reconstruct the sequence of pathological events in muscle morphology in FSHD, but overall a congenital component of the muscle pathology is probable. Extracellular matrix depositions and cellular infiltrates appear initially reversible. Their tipping points are subject of considerable attention. Animal models with provoked DUX4 expression, though none fully replicates the human disease, might help in finding clues to the earliest morphological and molecular changes. This is crucial in order to answer some of the key questions in the pathophysiology of FSHD.
Lessons learned from the molecular biology of FSHD
The molecular muscle biology of FSHD is said to involve many different pathways. As the sequential stages of pathology are still subject of discussion the stage-related molecular changes remain in doubt.
Expression of the transcription factor DUX4 in postnatal muscle has reportedly widespread and complex metabolic consequences. These downstream effects have been extensively reviewed recently [5, 39]. Various mechanisms enhancing DUX4 production have been reported. DUX4 expression leads to inhibition of nonsense-mediated decay [3, 19]. Accumulated transcripts, truncated proteins and unknown degradation products add to and complicate the pathogenesis of FSHD.
The present evidence is that DUX4 is expressed in brief bursts [5]. The trigger of these bursts is unknown. RNA and protein expression studies in cell-cultures and muscle biopsies have revealed the activation of a large range of intracellular and extracellular signals and pathways. Furthermore, chemokines, cytokines, myokines, MyoMir’s, and lncRNA’s have been detected in muscle biopsies and myoblast studies [5, 40]. Their function and possible interactions related to the various conditions of the diseased muscles are presently not clear.
The pathways can be grouped into clusters that relate to observed morphological changes [41]. Immunological and inflammatory pathways fit into the observations of cellular infiltrates possibly at the doorstep of necrotic cell death [33, 34]. Pathways activated in hypoxia and oxidative stress, in HIF-signaling and mitochondrial dysfunction could be grouped in the light of hypo-vascularity and the presence of lobulated fibers in the muscle biopsies [42–44]. Most interesting are the activated pathways of Wnt-beta-catenin signaling and myogenesis [45]. They might signify attempts at restoration of damaged muscle cells and explain the small fibers visibly as variation in fiber size in the muscle biopsies.
As DUX4 expression cannot be demonstrated directly in muscle biopsies its expression is presumed by the downstream effect: the activation of genes involved in early embryogenesis [41]. The zebrafish and Xenopus studies point to an overall toxicity of DUX4-mRNA in these specific studies. The studies on fetal human FSHD tissues suggest its expression is compatible with apparent normal body development [46]. This is also compatible with the myodevelopmental genesis of FSHD. While the unraveling of the diseased metabolic network of FSHD is likely to be a complex endeavor, the last two considerations bring us to the next paragraph that will explore congenital aspects of FSHD.
Observations pointing to FSHD harboring a congenital condition
The early muscle and systemic manifestations might point to a developmental or congenital component in FSHD. First, a possible effect on brain development in some individuals cannot be ruled out. Early onset FSHD distinguishes itself from the adult-onset patients by a more severe muscle weakness, more rapid progression and more frequently occurring systemic features. Some systemic features (cognitive disability, epilepsy) are only associated with early onset FSHD [47, 48]. The publications regarding these aspects is likely to be biased. A recent longitudinal study in children could not confirm these aspects being part of FSHD [49]. In our cross sectional study in 32 familial and 7 sporadic cases in 1995 we detected a mild to moderate retinal vasculopathy, consisting of retinal telangiectasis and microaneurysms in 18 of 37 evaluable angiograms (49%); five patients had minimal changes and 14 angiograms (38%) were normal. High frequency hearing loss was found in 25 (64%) out of 39 patients [50]. The non-progressive nature of retinal abnormalities is presumed based on similar findings in young and old individuals. More recently a longitudinal study confirmed its stationary nature [49]. The retinal vessel wall changes might be a result of altered (mesodermal) pericytes [51]. The retinal vasculopathy has a variable severity related to the size of the repeat [52]. So far, it is unclear whether the intra-muscular hypo-vascularity is a congenital condition or part of the later phase of the dystrophic pathology [33].
Pectus excavatum occurs more frequently in the FSHD population than in the normal population and is clearly part of the FSHD phenotype [53]. Similarly to facial weakness, it usually becomes visible in early childhood and is reportedly present in 5–16% of patients. It is considered to be a disorder of the costal and sternal cartilage-condition but molecular knowledge on its mechanism is not available [53].
A specific congenital hypoplastic myopathy is assumed but still needs proof. A number of recent studies support the hypothesis of a congenital muscle function defect. First, specific force, defined as the maximum voluntary contraction per contractile cross-sectional area of normal appearing muscle on MRI, is 33% reduced in FSHD compared to controls [54]. Single muscle fiber studies revealed a change of calcium sensitivity of force and an increase of passive force. Both were said to be related to an increased stiffness and titin content of the muscle fibers [55]. A recent study of the transcriptome of innervated FSHD1 and FSHD2 fibers suggests a sarcomeric dysfunction and reduced contractility [56]. Furthermore, genes for actin cytoskeleton signaling were found to be overexpressed in biceps muscles from patients compared to controls and deformed FSHD myotubes were demonstrated to have disorganized microtubule and actin networks [57]. All these studies were performed in cell cultures and diseased muscles. Whether muscle morphology and function is also abnormal in FSHD muscles that do not develop signs of muscular dystrophy during lifetime remains so far unanswered.
The sarcomere dysfunction summarized above might point to the presence of a mild congenital myopathy, microscopically visible as a variation in fiber size and clinically as the thin hypoplastic muscles of early onset patients [28]. This congenital myopathy could be a scaffold for further pathology as is proposed in the next paragraph.
Towards a hypothesis of the onset of FSHD pathogenesis
Explaining all aspects of FSHD has proven difficult, partly due to it being an exclusive human disease with imperfect animal models and partly since embryogenesis and fetal development in humans are challenging to study. Also, there is a lack of studies in minimally affected and non-penetrant cases, leaving the first stages of the disease in limbo. The absence of DUX4 in muscle biopsy studies makes the sequence of pathological development hard to reconstruct.
As mentioned, DUX4 is expressed in the germline and ZGA early in embryogenesis but is silenced after the 8-cell zygote stage [16]. DUX4 is expressed in fetal FSHD muscles but what happens afterwards is unclear [58]. The body plan develops normally and the muscle architecture, muscle positions and functions appear normal in early life. Formulated from a developmental perspective, DUX4 is effectively silenced in all endodermal and ectodermal tissues (with possible exception of the keratinocytes) [17]. As suggested by clinical features DUX4 is silenced in the facial muscles of some gene carriers with small deletions. In these patients DUX4 is often not expressed in lower extremity muscles based on clinical and MRI criteria. Similarly, in asymptomatic cases, even in those with large deletions, DUX4 expression must be reduced, and DUX4 must be stably silenced and apparently refractory to activation in non-penetrant cases and in the 2% of the general population carrying a reduced 4qA allele [9, 59].
These observations together suggest that the variable phenotype of FSHD reflects a lifelong attempt to keep the repeat silenced in almost all tissues and instances. At some point, with certain triggers, this attempt fails in muscles and FSHD becomes manifest.
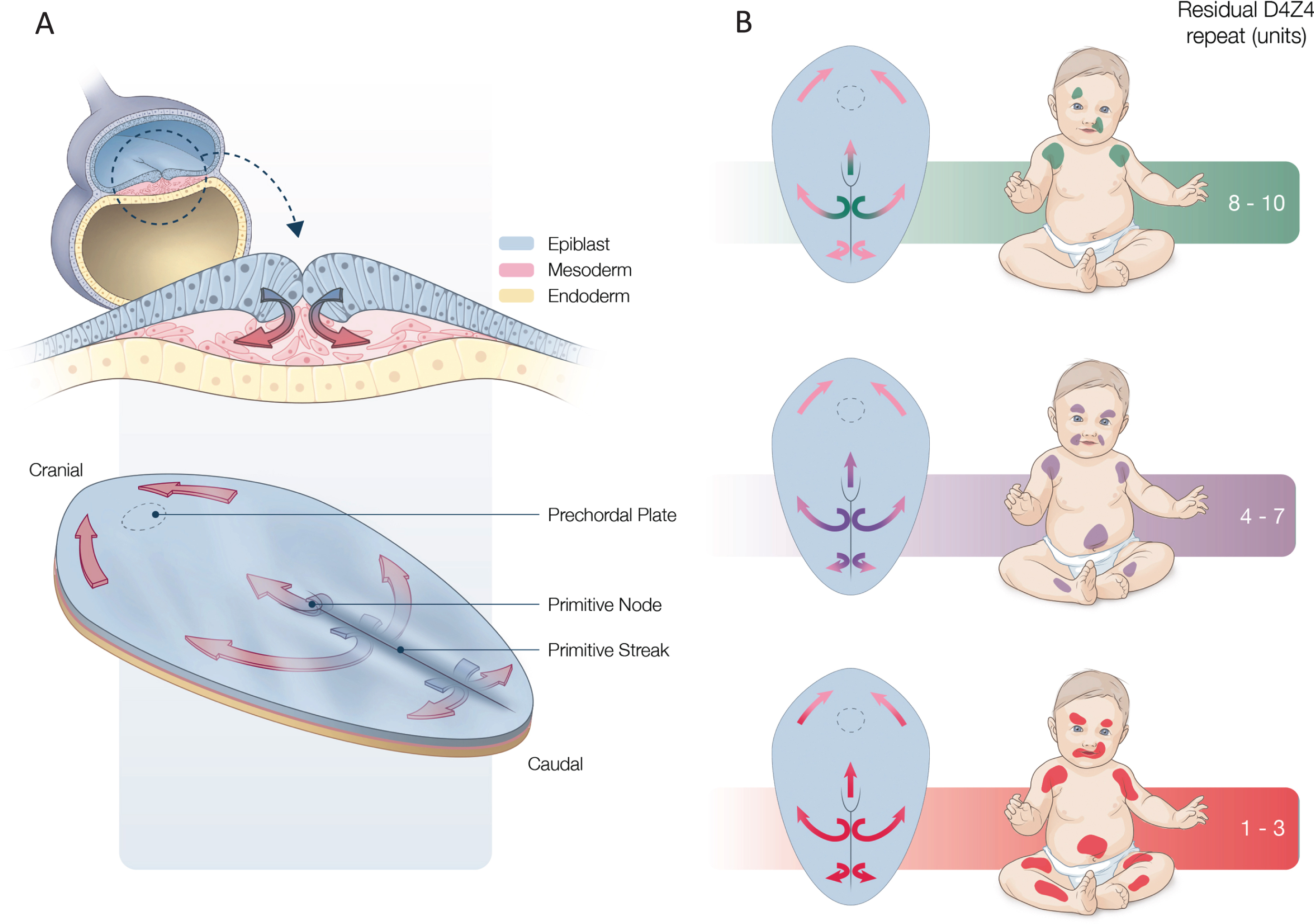
Graphical picture of the hypothesis, FSHD as a myodevelopmental disease. A: 16 days old embryo. The epiblast invaginates in the primitive streak and forms the mesoderm which extends in rostral, lateral and caudal direction. B: In FSHD a few early embryonic cells escape epigenetic silencing at the D4Z4 repeat. In endodermal and ectodermal development the silencing is accomplished as yet. In the mesoderm silencing is partial and incomplete improving with successive cell-divisions roughly related to the repeat-size. This leaves a rostrally, laterally and caudally decreasing gradient of weakly silenced stem cells. These cells develop in weakly silenced satellite-cells that will express DUX4 upon activation. Newborns carry thus a scaffold upon which FSHD becomes manifest. Adapted from: 1: Figure 3.3. from Larsen’s Human Embryology. 6th Edition - November 29, 2020.
Assuming a circulating or external agent as the trigger of FSHD onset requires a fate-plan or scaffold to work on. When all aspects of FSHD and all tissues involved are taken into account, this scaffold has to be laid down during the formation of the mesoderm [60]. The mesoderm is the first common ancestral tissue expressing features of FSHD later in life. This layer forms by invagination of the epiblast at the primitive streak from day 16 of embryonic development. In cranial direction it contributes to the prechordal plate and laterally it forms the paraxial, intermediate and lateral plate mesoderm. From day 18 onwards the paraxial mesoderm becomes organized into somitomeres and subsequently somites are formed in a rostro-caudal sequence over a period of several days (Fig. 4).
With this pattern in mind, one could speculate that in FSHD a few mesodermal cells escape silencing of the D4Z4 repeat early in embryogenesis. After each cell division the epigenetic landscape needs to be reconstituted. This landscape also changes over time as it drives development. All muscles are formed during fetal life by dividing muscle progenitor stem cells. Each division offers more cells an opportunity to silence the repeat as yet. Muscle progenitor cells divide asymmetrically [60]. Each progenitor cell produces a new progenitor cell and a number of muscle stem cells that differentiate into satellite cells. The mesoderm in FSHD is assumed to start out as a mix of weakly-silenced stem-cells (WS-SC’s) and normally silenced stem-cells (NS-SC’s). From this follows that in FSHD a rostro-caudal, medio-lateral and a dorso-ventral gradient of progressively better silenced stem-cells develops in the embryo. This leaves a gradient of WS-SC’s that peters out in caudal and lateral directions. As the number of mesodermal cells initially escaping silencing is assumed to be dependent on the number of residual repeats in the D4Z4 array, the extent of this gradient is also roughly dependent on it. The gradient is a mix of WS-SC’s and NS-SC’s in a composition wherein the amount of WS-SC’s at each level is roughly repeat-size dependent. The number of WS-SC’s translates into a gradient of vulnerability which becomes clinically apparent in a time dependent manner. The bisulfite sequencing data of the distal most D4Z4 repeat supports the idea of WS-SCs and NS-SCs as individuals actually show a wide range of methylation profiles for each individual chromosome assessed [61]. Thus assuming a basal triggering mechanism of pathology, which will be discussed below, the phenotype becomes a gradual progressive disorder descending downwards in the body.
A similar sequence of events could work out in a cranial direction for the development of the mesoderm of the prechordal plate [60, 62]. The result would be that the paraxial mesoderm of the second brachial arch and origin of the facial muscles is also populated with WS-SC’s and NS-SC’s. Their distribution is random and their number proportional to the residual repeat length. These WS-SC’s could leave some of the facial muscle fibers atrophic, hypoplastic and others fibers dead and absent. The facial muscles might be more vulnerable than others due to their specific transcriptional formation cascade [62, 63]. The anlage of other cranial muscles such as masseter and tongue might attract a different mix of stem-cells with fewer WS-SC’s so that only large D4Z4 deletions produce sufficient WS-SC’s to produce hypoplastic tongue and masseter muscles [29]. In other tissues of the cranial mesoderm, such as the retinal endothelium, a similar series of events could play out and lead to a disorganized pattern of tortuous and absent vessels. Incidentally, both facial muscle stem cells and retinal stem-cells use the same cytokine system, SDF1-CXCR4, for stem cell guidance [52, 63].
When the gradient of WS-SC’s is established in the truncal and limb anlage, an identical scenario for the skeletal muscle formation plays out. In the fetal phase of muscle growth myotubes expand into myofibers by adding proliferating MuSC’s. It is postulated that in the early development of the musculature of FSHD the gradient of WS-SC’s adds poorly functioning or atrophic stem-cells to the fibers creating the congenital myopathic scaffold. More importantly, apart from slightly weaker muscles in the gradient-zone, the gradient leaves a trail of WS-SC’s as satellite cells in their sublaminar niche, which adds another type of satellite cells to the heterogeneous pool in the niche [64]. In general stem-cells de-differentiate when activated [65]. We postulate a similar fate for muscle stem-cells. Subsequently, activated WS-SC’s in FSHD de-differentiate and express DUX4 which initiates pathology. It is assumed that in the formation of the embryological gradient the individual muscle anlagen in the trunk and limbs attract different numbers of WS-SC’s due to their different epigenetic addresses. This will result in the variable susceptibility to disease of individual muscles. This could explain the initial and relative sparing of the deltoid and supra- and infraspinatus muscles and also the relative early involvement of the semimembranosus and medial gastrocnemius muscles in the lower limbs [66]. In our experience, if patients manifest lower body weakness first, upper extremity weakness and atrophy is invariably present. Most likely, these patients have not experienced (repeated) high demands on upper extremity strength. We have shown that it takes asymptomatic patients many years before symptoms of shoulder girdle weakness are reported [10]. Furthermore, the strength of (upper extremity) muscles in the normal population show a wide variability (Brouwer et al. Brain 1992). Also, in the normal population asymmetries in the thorax and scapular position are not uncommon as is attested by the false positive clinical diagnoses in family studies [10]. We would not be surprised to find so-called non-penetrant gene carriers expressing subclinical FSHD when studied at the physiological, morphological or molecular level. By reciprocity, it might be possible that the 2% of carriers with a reduced D4Z4 repeat in the population contribute to the variability of upper extremity strength in the population. These assumptions define FSHD as a “satellite cell-opathy” and most likely as a primary satellite cell disease [67].
It is uncertain what happens in childhood, the phase in which muscle growth is moderate [31]. It appears that in most FSHD cases a tipping point is reached at the end of the childhood or in adolescence.
Vascular, infectious, immunological, toxicological, metabolic and oncological conditions do not play an important primary role in triggering FSHD in this life stage although they might contribute to some extent later in life [15]. This leaves trauma, ageing and degenerative processes as possible mechanisms. The curves of onset of the disease for larger deletions are ogives reaching plateau levels before the age of 50. This makes ageing and degeneration as exclusive causes less likely mechanisms [10]. Therefore we consider trauma a major pathogenic mechanism in FSHD, similar as in other muscular dystrophies. Nevertheless, it is still unsettled whether extreme muscle building and exercise might be beneficial or whether athletic activities in the teens might be the reason why specific gene carriers manifest symptoms and signs of FSHD in adolescence or adulthood.
Muscle as a mechanic organ is pre-eminently prone to physical stress [68, 69]. Particularly, eccentric contractions have been found to be extremely damaging to muscle fibers. In the upper extremities the scapula-fixators are almost constantly involved in eccentric contractions by use of the arms. Mild muscle damage could be restored by vesicular replacement without MuSC involvement [70]. More severe damage leads to plasmalemma rupture and cytosolic calcium influx. Loss of “moonlighting” enzymes activate MuSC’s and also the WS-SC’s in the niche, all of which is epigenetically regulated [71, 72]. Stem-cells in terminally differentiated tissues tend to de-differentiate for a short period when activated [65]. If this happens in FSHD WS-SC’s will express DUX4, which could explain the rare presence of DUX4 positive fibers in FSHD muscle biopsies.
When DUX4 expressing cells fuse to the damaged fiber, DUX4 production could be activated in neighboring myonuclei and be reinforced by several feedback-loops before being silenced again. DUX4 activates germ-line related pathways which, it is assumed, interfere with the complex process of restoring damaged myofibers.The massive influx of calcium in the cytosol in the presence of a possible poor vascularity pushes the metabolic stress towards the hypoxic state [33, 45]. This leads to energy depletion, acidosis (muscle pain) and the activation of fibro-adipogenic progenitor cells (FAP’s) [7, 51]. Cytokine and myokine production lead to an abnormal and inadequate inflammatory response. The latter has a characteristic composition at the cellular level and brings us to the recognizable morphological level of the microscope and the MRI scan. The infiltrates might be reversible initially, but the details of this process in FSHD have still to be learned [33, 36].
FAP’s are said not to express DUX4. They could be activated by neighboring cells with which they have ample means of communication. FAP’s express IL6, and IL6 was the only cytokine to be found chronically upregulated in FSHD [73, 74]. FAP’s are involved in many pathways balancing between restorative and pathological activities [75]. What triggers the balance to shift to pathology in FSHD is presently unknown. The FAP population is heterogeneous under normal conditions and the FSHD genotype could have added a new one [51, 76]. When activated it could lead to abnormal (deposition of) collagen and other extracellular matrix molecules, responsible for the early feature of fibrosis and cytokine activation in FSHD [7]. It could be similarly speculated that the sterno-costal collagen as a tissue of mesenchymal origin might not be of sufficient quality to resist the low intrathoracic pressures created by the diaphragmatic respiration resulting in a pectus excavatum.(92)
The proposed mesodermal gradient of WS-SC’s might also have influenced the development of the endomysial vasculature. If indeed the vascularity of diseased FSHD muscles is compromised [33] and is added to the natural proximal-distal decrease of muscle vascular efficiency [77], it might explain the characteristic distal to proximal progression of fatty infiltration in FSHD muscles [78].
DISCUSSION
The pathogenesis of FSHD has proved to be difficult to unravel and the genetic preconditions have been found complicated and are still not entirely clear. The proximity of the D4Z4 repeat, the 4qA allele with the PAS and the proximal SSLP on chromosome 4q35 allowed for a clinical autosomal dominant pattern of inheritance which led to the location of the gene by linkage analysis. Genetic diagnostics then identified non-penetrant cases. This observation solved the outstanding issue of anticipation as non-penetrance violates the criteria for anticipation. These cases also inspired to pay less attention to the progressive muscle pathology but more on ideas how FSHD muscle could escape the activation and expression of DUX4.
The present hypothesis describes a rostro-caudally declining number of epigenetically WS-SC’s in the FSHD embryo. They are activated in adult muscles to de-differentiation in damaged muscle fibers and produce DUX4. Dependent on the amount of WS-SC’s and the extent to where they were able to spread during development a progressive muscle phenotype might develop in adult life. This sequence of events offers explanations for some of the enigmatic features of FSHD; It helps to explain the development of the characteristic phenotype in a variable rate over years; The epigenetic addresses directing stem cells to developing muscles might offer explanations for the relative sparing of some muscles early in the course of the disease and for some preferred muscle involvement in the lower limbs. It must be assumed that in FSHD some epigenetic addresses attract more WS-SC’s to their destination than others; A similar epigenetic explanation might be applicable to the often asymmetrical affection of the facial and skeletal musculature; The FSHD phenotype reveals several gender differences. The age at onset is generally reported to be later in women than in men. There are more asymptomatic females with mild disease than asymptomatic males in completely examined families [10, 11]. Female mosaics were reported to have a higher tolerance to DUX4, as similar proportions of mutated cells with identical numbers of residual repeats resulted in milder signs of the disease compared to men [13]. Explanations are often sought in estrogen effects but a recent study on epigenetic signatures showed that in women muscles are generally more methylated at the repressive histone H3K9 marker than in men [79]. This might have consequences for the degree of repression of the D4Z4 repeat in muscle stem-cells and contribute to the apparent increased tolerance of women to the toxic effects of DUX4; Cardiac abnormalities in FSHD are usually mild, consisting of an increased frequencies of a left sided bundle branch block or conduction abnormalities in elderly patients compared to controls [80]. An explanation is often sought in the embryonic origin of a part of the left myocardium in the second brachial arch, which is the same tissue area as facial muscle anlage; Little is known about the pathology of the inner ear in FSHD to explain the high tone hearing loss. As this loss resembles effects of noise trauma and presbycusis, the usual explanation is sought in involvement of the stapedius muscle innervated by the facial nerve. Normally this muscle is involved in a reflex pathway drawing the stapes from the footplate in case of loud noise, protecting auditory sensory neurons from damage. Ageing it is said offers an additional effect. However, research has not excluded a neurogenic contribution to the deafness; There are three symptoms related to the muscle condition that do not follow directly from the mesodermal discussion. However they are important consequences of the disease and must be explained by downstream effects of DUX4 expression. Stiffness, pain and fatigue are largely subjective and are usually approached by questionnaires. They need better molecular underpinnings to see whether they are specific symptoms for FSHD or general symptoms for muscle disease.
Muscle stiffness has rarely been studied and was reported to be present in 60% of FSHD patients [81]. This symptom was explained by extracellular matrix and collagen deposition; tendons or joints were not discussed or mentioned [51]. Muscle pain is reported in up to 90% of all patients. However, studies on muscle acidosis, free radical production, cytokine and chemokine production related to pain in FSHD are difficult to find [40, 83]. Also, its frequently chronic nature has been neglected. Central sensitization has not been studied; brain involvement in FSHD is still an open question [84]. Finally, the high frequency of fatigue in FSHD has received considerable attention. It has a firm basis in the muscle condition. It needs to be determined whether the effects of training and cognitive therapy are specific for FSHD as well [85]. Persons who never suffer these complaints might need more study.
EPILOGUE AND CONCLUSIONS
FSHD is probably the most prevalent muscular dystrophy. Its prevalence rate is estimated 1:8300 using a capture-recapture method and three independent registries in the Netherlands [86]. As it is unlikely that these registries harbor large numbers of asymptomatic and non-penetrant cases, the true prevalence might be at least 30% higher. The non-penetrant cases of the FSHD families add to the number of persons with a shortened 4qA allele (2%) in the normal population. This addition can easily be counterbalanced by a postulated mutation frequency of gene-crossover with expansion of the repeat in the non-penetrant population [14, 88], fulfilling criteria for a Hardy-Weinberg Equilibrium.
The only premise of our hypothesis, which would fit Ockham’s requirements, is the existence of a specific stem cell epigenetically weakly repressed at the D4Z4 repeat [67]. We propose that FSHD is a myodevelopmental disease in which a few embryonic cells escape epigenetic silencing, related to the D4Z4 repeat size, which results in a fetus with a gradient of mis-regulated satellite cells leading to an abnormal myofibrillar structure that is the key mis-formed scaffold resulting in FSHD phenotype. This hypothesis has great consequences for both prognosis and therapy. The embryological part of the hypothesis might be difficult to proof. It consequences, however, appear all falsifiable. If the extent of the congenital condition can be diagnosed in patients at the earliest signs of the disease, it will aid in directing therapeutic strategies.
Footnotes
ACKNOWLEDGMENTS
The authors of this publication are members of the Radboudumc Center of Expertise for neuromuscular disorders (Radboud-NMD), Netherlands Neuromuscular Center (NL-NMD) and the European Reference Network for rare neuromuscular diseases (EURO-NMD). No funding was received for this project.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.