
Abstract
Emery-Dreifuss Muscular Dystrophy (EDMD) is an early-onset, slowly-progressive group of myopathies, presenting with joint contractures, muscle weakness and cardiac abnormalities. Variants in the EMD gene cause an X-linked recessive form (EDMD1). The scarce EDMD1 muscle MRI accounts in the literature describe fatty replacement of posterior thigh and leg muscles.
We report a 22-year-old patient with early-onset bilateral joint contractures, slowly progressive muscle weakness and minor cardiac rhythm abnormalities. A novel loss-of-function variant of EMD was identified and deemed probably pathogenic in the absence of emerin detection by immunofluorescence and Western Blot. MRI revealed fatty replacement of the lumbar spinal erectors and the posterior compartment of lower limbs. Interestingly, Short Tau Inversion Recovery (STIR) sequences showed a heterogenous hyper signal on the vasti, hamstrings and left lateral gastrocnemius muscles.
Oedema-like abnormalities were previously reported in early stages of other muscular dystrophies, preceding fatty replacement and muscle atrophy, but not in EDMD1 patients. We hypothesize that these oedema-like changes may be a marker of early muscle pathology in EDMD1. Further studies focusing on these abnormalities in the early phase of EDMD1 are required to test our hypothesis.
ABBREVIATIONS
Creatine kinase
Emery-Dreifuss Muscular Dystrophy
Magnetic resonance imaging
Short Tau Inversion Recovery
INTRODUCTION
Emery-Dreifuss Muscular Dystrophy (EDMD) encompasses a group of genetically heterogeneous slowly progressive myopathies characterized by a similar phenotype. Classically, EDMD presents with a triad of early contractures, progressive muscle weakness and atrophy, and cardiomyopathy with conduction defect [1, 2]. Onset is usually in the first decade of life [3].
Variants in the EMD gene, which encodes emerin, a nuclear envelope protein, result in X-linked recessive inheritance myopathy (EDMD1). EDMD2 and EDMD3 correspond to the autosomal dominant and recessive forms, respectively, caused by variants of LMNA (encoding lamin A/C). Together, mutations of EMD and LMNA are the most frequently found causes of EDMD, representing about 40% of cases. Other genes have been associated with EDMD, namely SYNE1 (EDMD4), SYNE2 (EDMD5), FHL1 (EDMD6), TMEM43 (EDMD7), SUN1, SUN2 and TTN [1].
Muscle imaging has emerged in the last two decades as a valuable tool in the workup of muscle diseases. Magnetic resonance imaging (MRI) is the method of choice, thanks to its safety and excellent soft tissue resolution. It allows detection of muscle atrophy and signal changes, either from fatty replacement or oedema. Additionally, it is possible to single out the affected muscle(s). Specific patterns of muscle involvement have been described for several forms of myopathies [4]. The recognition of these patterns is important, especially in the context of next-generation sequencing, which may identify numerous variants in a single patient.
In EDMD, either due to EMD or LMNA variants, there is a characteristic fatty replacement of quadriceps, posterior thigh and leg muscles, while rectus femoris, gracilis and sartorius are relatively spared [5–7]. The degree of fatty replacement appears to be variable in peroneus muscles, being more frequent and severe in EDMD1 than in EDMD2 [6]. However, there are proportionally fewer reports on patients with EDMD1, and the pattern of muscle changes is seldom described.
We report on an EDMD1 case exhibiting peculiar Short Tau Inversion Recovery (STIR) hyperintensities on muscle imaging and harboring a novel, potentially pathogenic variant of the EMD gene.
CASE REPORT
A 22-year-old male was evaluated for a slowly progressive myopathy with contractures. He was born at term to non-consanguineous parents after an uncomplicated pregnancy and delivery. He achieved normal motor and cognitive milestones at the expected age. He practiced sports through childhood and adolescence. However, retrospectively, parents reported frequent falls during childhood. He had been otherwise healthy. There was no known family history suggestive of neuromuscular disorders.
When first examined at 7 years old, limited bilateral elbow extension and toe walking were noticed. By 11 years of age, clear bilateral elbow and ankle contractures were apparent, followed by the occurrence of scoliosis at 16 years of age.
On his last appointment, at 22 years old, he was fully ambulatory but could no longer play sports due to limb contractures. He reported a single episode of heart palpitations, with no other associated symptoms. He denied exercise intolerance, fatigue or pain. He had no sleep disorders. He displayed a toe-walking, waddling gait, and was unable to walk on his heels. On general examination, he presented mild bilateral winged scapula, rigid spine, lumbar lordosis and bilateral contractures involving the elbow, wrist, finger flexors, knee and ankles. Atrophy of dorsal and lumbar paravertebral muscles and both anterior and posterior compartments of upper and lower limbs was apparent. There was no cranial nerve involvement. No skin changes were detected. His cognition was normal. Motor examination (using the Medical Research Council score system) was rated as follows: neck flexion (3/5), shoulder abduction (4+/5), elbow flexion and extension (4+/5), finger extension (4/5), finger abduction (4+/5), hip extension (4/5), hip adduction (4+/5), hip flexion (5-/5), ankle dorsiflexion (4+/5) and toe extensors (3+/5). He had generalized areflexia.
The laboratory tests were unremarkable, except for moderately elevated creatine kinase (CK < 10x upper limit of normal, maximum 1795U/L) and mildly elevated AST and ALT (<3x upper limit of normal). The cardiac workup revealed sinus bradycardia and left anterior bundle block. Echocardiography was normal with a left fraction ejection of 65%. Electromyography showed reduced and polyphasic motor unit action potential with early recruitment. Left deltoid muscle biopsy (Fig. 1, panel I) displayed variable muscle fiber size, internal nuclei, and proliferation of connective tissue, with no inflammatory infiltrate. HLA1 immunostaining was negative. Immunofluorescence proved the absence of emerin expression. The same pattern was found in muscle Western Blot analysis.
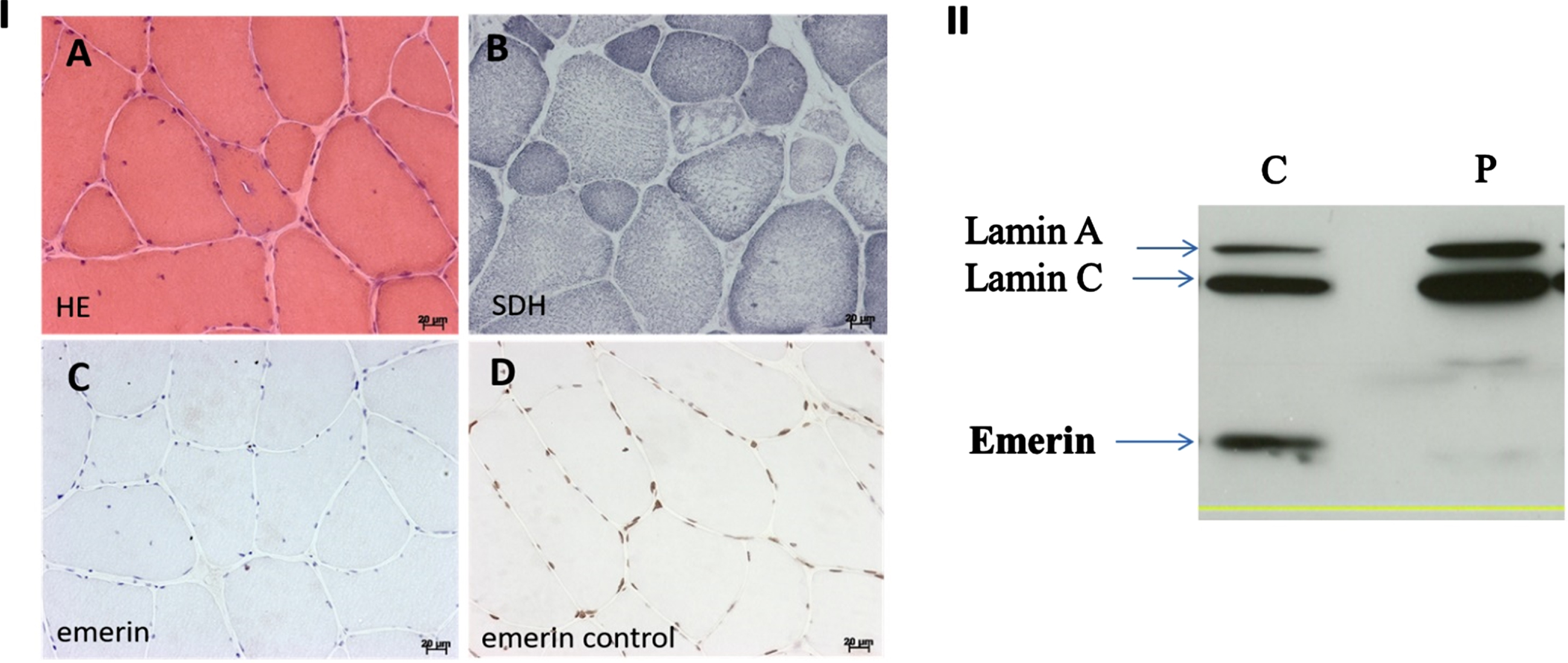
Panel I: Patient’s muscle histology. A (Hematoxylin and Eosin, HE): Variability in the size of muscle fibers, internalized nuclei, marked increase in endomysial and perimysium connective tissue. Few fibers contain a bordered vacuole. B (Succinate Dehydrogenase, SDH): Disorganization of the structure in some muscle fibers. C: Immune cytochemistry with an antibody directed against the emerin shows immune labelling absence in the nuclei. D: Control’s normal emerin labelling in the nuclei (nuclei appeared in brown). Panel II: Western-blot analysis of Emerin and Lamin on lymphoblastoid cells, using Manem8 Clone 7B9 Antibodies for Emerin and Manlac14A7 for Lamins. C: Control’s lymphoblasts; P: Patient’s lymphoblasts.
Initial lower limb muscle MRI (Fig. 2, panels A-D), performed at 19 years of age, revealed fatty infiltration (T1-weighted images) in the following muscles, graded as follows using the Mercuri scale [8, 9]: 4 for lumbar spinal erectors and medial gastrocnemii; 3 for the solei muscles and 1 + for the semimembranosus, biceps femori and lateral peroneus muscles. STIR sequences showed a heterogenous hypersignal on the vastii, hamstrings and left lateral gastrocnemius. This pattern of muscle involvement persisted in the whole-body muscle MRI, performed three years later (Fig. 2, panels E-H). Additionally, at that time, gastrocnemius atrophy (bilateral medial gastrocnemius and left lateral gastrocnemius) became apparent. Interestingly, these T1 and STIR hyperintensities were not observed in the upper limbs.
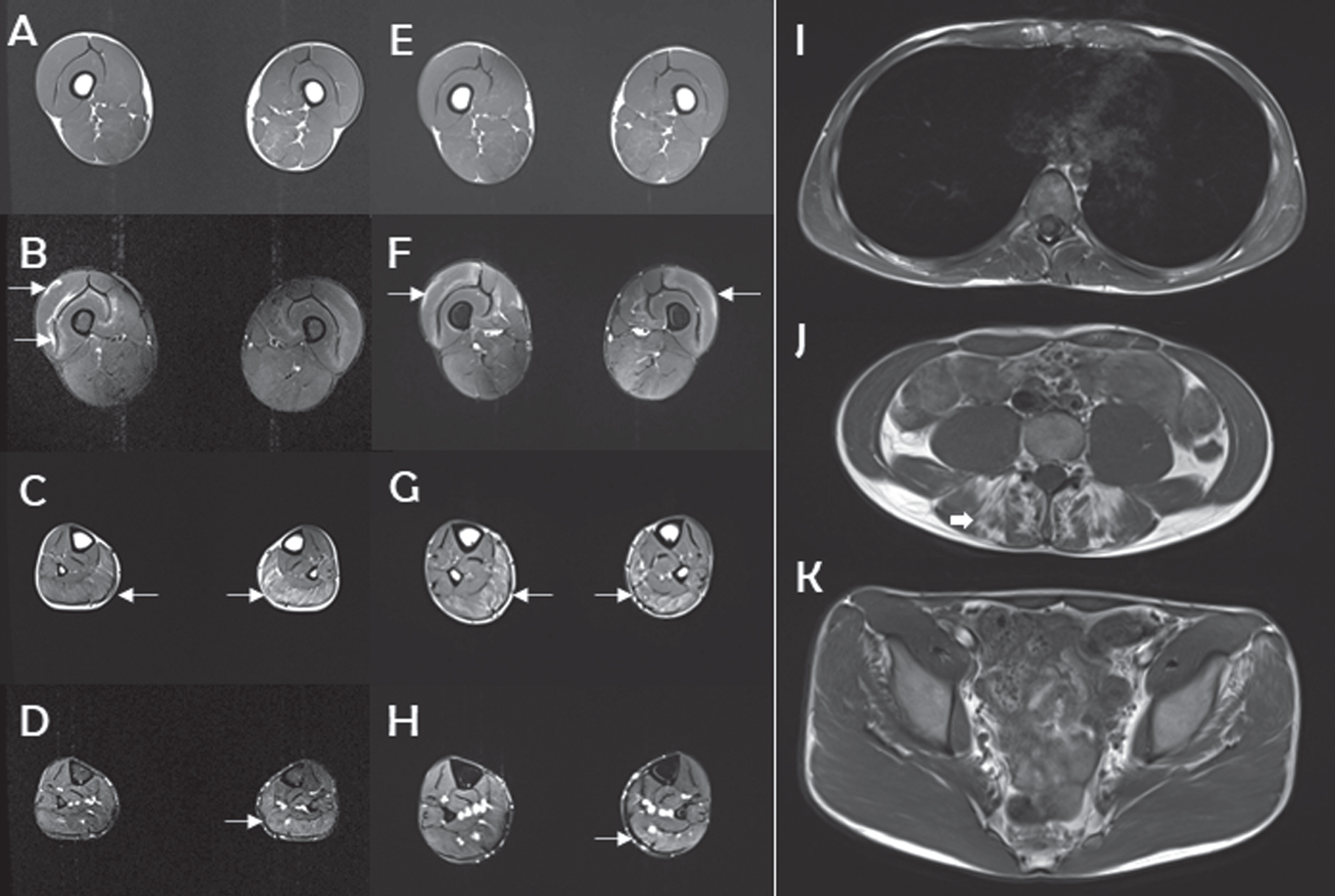
Representative features as revealed by muscle MRI imaging. Panels A-D: Lower limb muscle MRI performed in 2018 (19 years old). Panels E-H: Lower limb muscle MRI performed in 2021 (22 years old). Panels I-K: Dorsal, lumbar and pelvic level MRI in 2021 (22 years old). At the first visit (19 years old), T1 sequences showed no significant pathological changes in the thigh and focal fatty replacement of gastrocnemius muscles (C, arrows). These latter lesions show little evolution at follow-up (22 years old) (G, arrows). T2-STIR encoded sequences demonstrate focally enhanced lesions in the quadriceps (B, arrows) and left gastrocnemius muscles (D, arrow). These lesions appeared rather stable over time (F, H, arrows). T1-weighted imaging shows no muscle alterations at the dorsal and pelvic level (I, K), whereas lumbar erector muscles demonstrate clear fatty replacement (J, arrow). No signal abnormalities were found in the upper limb muscles.
A limb-girdle muscular dystrophy Next Generation Sequencing panel revealed a new variant of the EMD gene, NM_000117.2: c.83-1G>T (IVS1-1G>T), which was later confirmed by Sanger sequencing. This splice variant was absent in GnomAD [10], Clinvar [11] or LOVD [12] and it is predicted to alter splicing (98.4% by Splicing Prediction Pipeline, SPiP). No variants were found in the LMNA gene. The absence of emerin expression in both muscle biopsy and lymphoblasts further confirmed its pathogenicity (Fig. 1, panel II). The patient’s mother was found to be an asymptomatic carrier of the same variant, and his younger brother, who had no muscle symptoms or signs, did not carry the variant.
DISCUSSION
Our patient presented with early-onset joint contractures and slowly progressive muscle weakness, initially in a humeroperoneal pattern, then progressing to an axial and limb-girdle distribution, consistent with previous EDMD1 reports [1, 2].
Although the present EMD variant was not previously described, it was considered pathogenic owing to its predicted role in splicing through a loss-of-function mechanism. Nonsense variants represent the majority of known pathogenic variants in EDMD1 [3].
Descriptions of whole-body or lower limbs MRI in EDMD1 patients are scarce. Previous reports described the fatty replacement of tongue, trapezius, biceps brachialis, paravertebral, glutei, quadriceps, hamstrings, adductor major, solei and gastrocnemius muscles [1, 13]. Rectus femoris, gracilis and sartorius seem to be more preserved. The fatty infiltration mainly affects the posterior compartment of both thighs and legs [5–7]. Involvement of peroneus muscles seems to be of particular importance in distinguishing the two most common forms of EDMD, as they were severely affected in 88% of EDMD1 patients compared to only 40% of patients with LMNA variants in a Spanish cohort [6].
In our patient, lumbar paravertebral, posterior leg and, to a minor degree, posterior thigh muscles were damaged, evidencing fatty replacement. Muscle involvement was mainly symmetric, but focal mild asymmetry was found in the gastrocnemius muscles, as described in other reports [6].
Oedema-like abnormalities seem to be a precursor of fatty replacement and muscle atrophy in other dystrophies, such as Duchenne, myotonic and facioscapulohumeral dystrophies [14–16]. Degardin et al. [17] reported that hyperintensities observed on STIR sequences precede fatty degeneration in 62% of their muscular dystrophies cohort. Marden et al. [18] described muscle oedema in the thighs of young patients with dystrophinopathies. Another study focusing on limb-girdle muscular dystrophies also reported the detection of oedema-like lesions as the first step in muscle disease, before fibroadipose changes [19]. Oedema-like changes were also found in Miyoshi myopathy and young Pompe disease patients’ thigh and calf muscles [20, 21]. However, there are currently no similar reports in EDMD patients.
On the contrary, we did not find reports on STIR abnormalities found in non-dystrophic myopathies, including collagenopathies and congenital myopathies [22]. Therefore, we hypothesize that this oedema-like pattern may be found in muscular dystrophies, particularly in lower limbs, but not in non-dystrophic myopathies, such as congenital myopathies.
Interestingly, the pattern of hypersignal observed on T2-weighted and STIR sequences in our patient was markedly heterogeneous, localized on the quadriceps and left gastrocnemius muscles, and, to our knowledge, not previously described in EDMD1. We hypothesize that it may correspond to an early phase of muscle involvement by the disease prior to fatty replacement. Of note, no correlation of these MRI hyperintensities was noticed with muscle strength. As a matter of fact, the upper limb muscles displayed no T1, T2-weighted or STIR abnormalities, despite the clinically apparent weakness. Carboni et al. [7] had already reported an apparent mismatch between the presence and severity of muscle weakness and muscle imaging abnormalities. In a family of 5 males harboring an EMD variant, all depicted MRI abnormalities of posterior leg muscles, yet only two of them displayed muscle weakness on clinical examination.
Whether the hyperintensities in STIR result from inflammation or other pathogenic process remains debatable. EDMD seems to be caused by a defect in the importation of nuclear envelope proteins into the nucleus, probably leading to the loss of its structural integrity. Mutant forms of emerin were associated with decreased nuclear invagination and abnormal calcium transient [1, 23] Cardiac and skeletal muscles are at particular risk, owing to their high energy demand. Impaired differentiation of myogenic progenitors and inefficient muscle cell regeneration appear to be the result [24]. It is not currently known if this genetic defect might trigger chronic inflammation. There are some reports of macrophage-mediated inflammation in LMNA-related myopathies 25, 26], nevertheless, to our knowledge, no such studies were performed in EMD-related myopathies. Additionally, in LMNA myopathies, inflammatory cells on muscle samples seem to be more numerous in young than older patients [25], which may reinforce the possible role of inflammation in the early stages of the disease. In our patient, the muscle biopsy showed no inflammatory infiltrate, but it was performed in the deltoid muscle, which showed no sign of impairment on the whole-body MRI. Hence, we cannot exclude the possibility of an early inflammatory insult in EDMD1 solely based on the results of muscle histology.
In conclusion, our patient, who harbors a novel potentially pathogenic variant of the EMD gene, displayed oedema-like abnormalities on STIR, as described in the early stages of other dystrophies, before the development of fatty degeneration. It is possible that oedema-like changes may represent a marker of early disease. Further studies focusing on the possible role of inflammation and oedema in the early phase of EDMD1 are required to test our hypothesis. Longitudinal muscle MRI of EDMD1 patients is required to understand the fate of these T2-weighted and STIR hyperintensities.
FUNDING
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
DECLARATIONS OF INTEREST
None.