
Abstract
Introduction:
Nuclear envelopathies occur due to structural and/or functional defects in various nuclear envelope proteins such as lamin A/C and lamin related proteins. This study is the first report on the phenotype-genotype patterns of nuclear envelopathy-related muscular dystrophies from India.
Methods:
In this retrospective study, we have described patients with genetically confirmed muscular dystrophy associated with nuclear envelopathy. Data on clinical, laboratory findings and muscle MRI were collected.
Results:
Sixteen patients were included with median age at onset of 3 years (range: 1 month – 17 years). Three genes were involved: LMNA (11, 68.75%), EMD (4, 25%) and SYNE1 (1, 6.25%). The 11 patients with LMNA variants were Congenital muscular dystrophy (MDCL)=4, Limb Girdle Muscular Dystrophy (LGMD1B)=4 and Emery-Dreifuss Muscular Dystrophy (EDMD2)=3. On muscle biopsy, one patient from each laminopathy phenotype (n = 3) revealed focal perivascular inflammatory infiltrate. Other notable features were ophthalmoparesis in one and facial weakness in one. None had cardiac involvement. Patients with EDMD1 had both upper (UL) and lower limb (LL) proximo-distal weakness. Cardiac rhythm disturbances such as sick sinus syndrome and atrial arrhythmias were noted in two patients with EDMD1. Only one patient with variant c.654_658dup (EMD) lost ambulation in the 3rd decade, 18 years after disease onset. Two had finger contractures with EMD and SYNE1 variants respectively. All patients with LMNA and SYNE1 variants were ambulant at the time of evaluation. Mean duration of illness (years) was 11.6±13 (MDCL), 3.2±1.0 (EDMD2), 10.4±12.8 (LGMD1B), 11.8±8.4 (EDMD1) and 3 (EDMD4). One patient had a novel SYNE1 mutation (c.22472dupA, exon 123) and presented with UL phenotype and prominent finger and wrist contractures.
Conclusion:
The salient features included ophthalmoparesis and facial weakness in LMNA, prominent finger contractures in EMD and SYNE1 and upper limb phenotype with the novel pathogenic variant in SYNE1.
INTRODUCTION
Nuclear envelopathies are a heterogenous group of disorders due to defects in the protein coding genes of nuclear envelope which most commonly include Lamin A/C and Emerin [1]. There are varied neuromuscular manifestations such as muscular dystrophy, cardiomyopathy and inherited neuropathies. The skeletal muscle involvement has a wide spectrum of clinical manifestations ranging from congenital muscular dystrophies to adult onset phenotypes. Emery-Dreifuss Muscular Dystrophy (EDMD) is a genetically heterogenous group of hereditary muscular dystrophies. It is a rare disorder with an estimated incidence of 3 in 100,0000 population [2]. The classical triad of EDMD includes joint contractures, progressive muscle weakness with wasting and cardiac involvement. The most common pattern of weakness is the humero-peroneal form involving biceps, triceps and peroneal muscles with scapular winging and sparing of facial muscles [3]. EDMD is further classified as Emerin (EDMD1 – OMIM 310300), Lamin A/C (EDMD2 – OMIM 181350/EDMD3 – OMIM 616516), Nesprin-1 (EDMD4 – OMIM 612998) [4]. Other phenotypes described in laminopathies include congenital muscular dystrophy (MDCL – OMIM 613205) and proximal limb-girdle pattern (LGMD1B – OMIM 159001) [5]. There are no previous studies on the clinical and genetic findings of nuclear envelopathies related muscular dystrophies from India. Thus, this study aims to describe the phenotype-genotype heterogeneity of an Indian cohort.
METHODS
This is a retrospective descriptive study done on genetically confirmed nuclear envelopathy patients with muscular dystrophies from a quaternary neurology referral centre in southern India. Detailed clinical features including onset of symptoms, pattern of muscle involvement, progression, family history, laboratory features such as serum creatine kinase (CK) levels; muscle magnetic resonance imaging (MRI) graded for fatty infiltration using modified Mercuri grading [6] and muscle biopsy findings on light microscopy histopathology (eosin and hematoxylin) and myosin ATPase staining was obtained. Genetic analysis was done by next generation sequencing in all patients as previously described [7] (Supplementary Table 1). Identified variants were classified as pathogenic/likely pathogenic as per American College of Medical Genetics (ACMG) criteria using Franklin variant classification tool (https://franklin.genoox.com/clinical-db/home) [8]. Institutional ethics committee approval was obtained [IEC no: NIMH/DO/(BS&NS) 2022]. Informed consent was obtained from all the patients for publication of clinical data and images. Descriptive statistics such as mean, median and range were used to describe the data.
RESULTS
Sixteen patients were included with a median age at onset of 3 years (range: 1month – 17 years). The median duration of illness was 4 years (range: 1–31years). The male to female ratio was 4.6:1. Three genes were involved including LMNA (11, 68.8%), EMD (4, 25%) and SYNE1 (1, 6.3%). Other genes associated with EDMD such as SYNE2, TMEM43 and FHL1 were not detected in our cohort.
(Phenotype and genotype details of cohort in Table 1)
Clinico-genetic details of the patients
EMERIN (EMD)
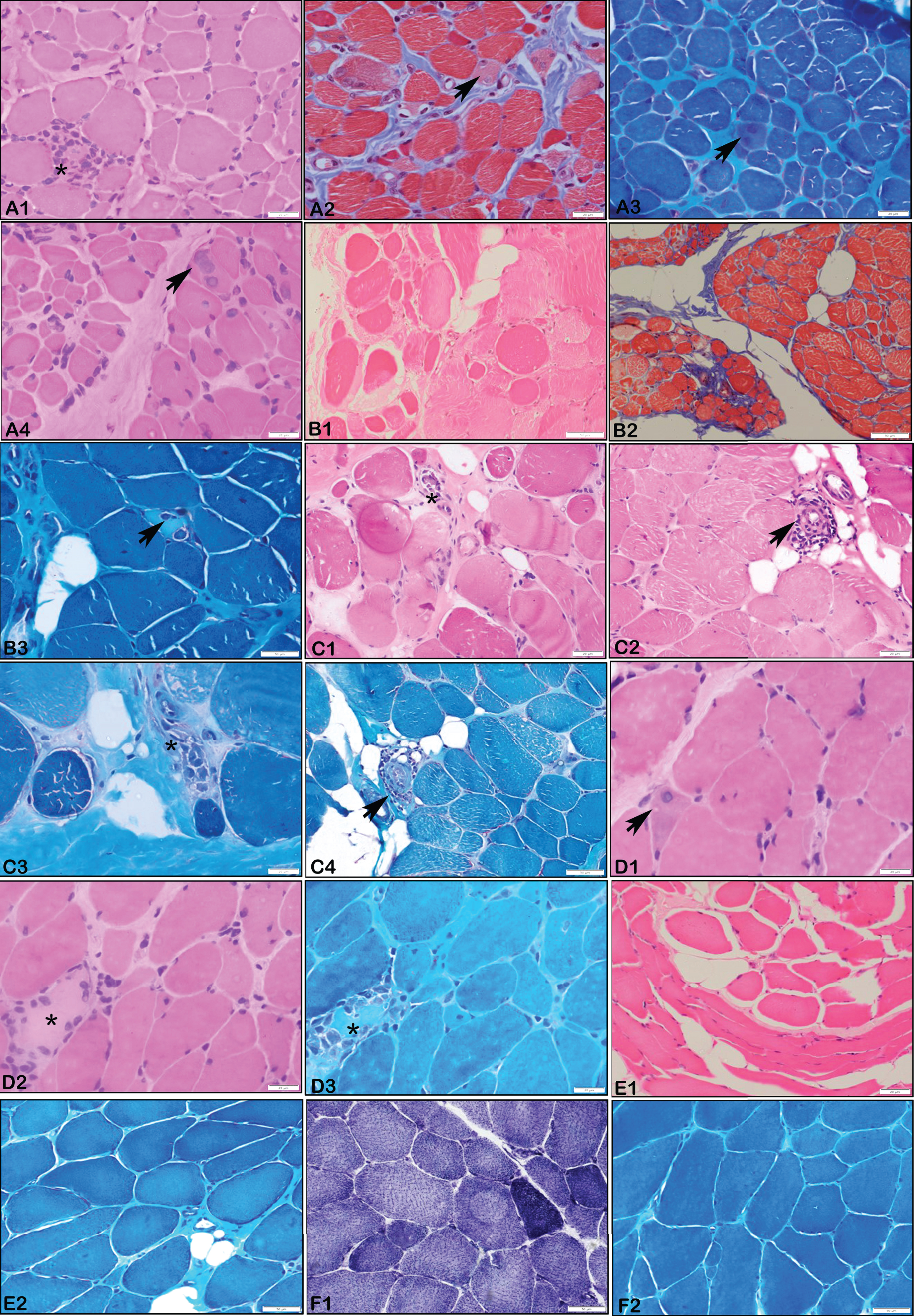
There were 4 patients with EMD pathogenic variants with phenotypic presentation of EDMD1. The median age at onset of symptoms was 3 years (range: 1 – 8 years) with varying duration of presentation ranging from 4 to 20 years. All patients were males. Consanguinity and positive family history of sudden cardiac death in first cousin was noted in P14. Both upper (UL) and lower limb (LL) weakness were noted in all patients. Truncal weakness was noted in only one. Only one patient (P12) lost ambulation and required wheel chair assistance at 26 years of age (18 years after disease onset). Cardiac involvement was noted in two patients– P12: sick sinus syndrome and left ventricular hypertrophy with implanted pacemaker and P13: atrial arrythmias. Talipes equino-varus deformity was noted in two patients (P12, P13) and lumbar lordosis in one (P15). All had ankle contractures with additional elbow, hip and finger contractures in P13 and posterior cervical contractures in P14. However, patients P12 (onset at 8 years) and P15 (onset at 3years) had atypical pattern with only ankle contractures at presentation. The median CK level was 894 U/L (range: 180–1371 U/L). Muscle biopsy was done in only one patient (P13) showing neurogenic changes with fibre type II grouping (Fig. 1F). All four males had X-linked recessive disorders with truncating hemizygous variations identified in EMD gene with exon 6 involved in three out of four. While two (P13, P14) have previously reported nonsense and frameshift variations respectively, P12 and P15 have frameshift variations identified for the first time in this study (Table 1). Notably both had novel pathogenic variants of c.654_658dup (P12) and c.350del(P15).
LAMIN-A/C (LMNA)
Of the 11 patients with LMNA pathogenic variants, 4 (P1, P2, P6, P7) had MDCL, 4 (P4, P9, P10, P11) had LGMD1B and 3 (P3, P5, P8) had EDMD2 phenotypes. Positive family history was noted in one (P11) and consanguinity in one (P4).
All patients with MDCL phenotype had onset of weakness at 1 year of age except P2 who had at 1 month of age. Developmental delay followed by limb girdle weakness was noted in all MDCL patients with associated distal LL weakness in P2. None of these patients had contractures at presentation. Other notable features were ophthalmoparesis in P7 and hyperlordosis in P6. Muscle biopsy done in P6 and P7 showed myopathic pattern with focal perivascular inflammation in P7 (Fig. 1).
Patients with LGMD1B phenotype had a median age at onset of 6 years (range: 2.5–17years). The pattern of weakness was proximal UL and LL (P9, P10), proximal LL (P11) and distal LL (P4). Other features such as hyperlordosis (P10, P11) and scapular winging (P9) were also noted. Contractures at ankles was seen in only two subjects (P9, P10). Muscle biopsy was done in P9 and showed myopathic changes with perivascular inflammation (Fig. 1).
The age at onset in EDMD2 ranged from 2 – 3.5 years. Except P8, all had proximal UL with proximal and distal LL weakness. Facial weakness (P5), scapular winging (P3, P5) and kyphoscoliosis (P5) were other features. Muscle biopsy done in two cases (P3, P8) showed myopathic features with myonecrosis and perivascular inflammation in P8.
Mean duration of illness (years) are 11.6±13 (MDCL), 3.2±1.0 (EDMD2), 10.4±12.8 (LGMD1B). All patients were ambulant at the time of evaluation. Median serum CK level was 860 U/L (range: 134–2603). None had cardiac symptoms and electrocardiogram and 2D echocardiography were normal in all probands. Muscle MRI done in two showed global fatty infiltration of thigh (P1, P11) and leg muscles (P11). All patients except P11 were sporadic and were found to have heterozygous variations in LMNA, with the most common variations being missense involving exon 1 (n = 3). One duplication variant at exon 10 causing frameshift (c.1527dup, p.Thr510TyrfsTer42) was noted in P11 (Table 1).
NESPRIN-1/SPECTRIN REPEAT CONTAINING NUCLEAR ENVELOPE PROTEIN-1/(SYNE1)
Patient P16 had onset of symptoms at 15 years of age and presented at 18 years. The proband had atypical phenotype with proximal and distal UL weakness without LL weakness. Prominent finger and wrist flexion contractures were noted. There were no cardiac symptoms, truncal or bulbar weakness. Elevated serum CK of 945 U/L was present. P16 had a novel homozygous frameshift variation affecting exon 123 of SYNE1 (Table 1).
DISCUSSION
Here we have described in detail the phenotype – genotype characteristics of 16 patients with envelopathies which result from structural and/or functional defects in the various nuclear envelope proteins.
Emery-Dreifuss muscular dystrophy
EDMD due to emerin and lamin A/C defects contribute to 40% of EDMD cases. Lamin A/C encoded by LMNA gene is the major component of nuclear membrane localised to the inner nuclear membrane and nucleoplasm [9]. Pathogenic variants in LMNA have a broad spectrum of heterogenous manifestations including congenital muscular dystrophy, autosomal dominant dilated cardiomyopathy with conduction defect, familial partial lipodystrophy and autosomal recessive Charcot Marie Tooth disease and progeria [10]. Lamin A/C binds with other proteins such as emerin (EMD), lamin- A-associated polypeptide (LAP) and MAN1 [11]. It plays a major role in chromatin organisation, replication, cell cycle regulation and structural stability to mechanical stress especially in tissues such as skeletal and cardiac muscles [12]. The estimated prevalence of all types of EDMD is about 1.3:100,000 – 2:100,000 [13]. There is significant inter- and intrafamilial variability in age of onset, progression and severity of muscle and cardiac manifestations. The variability in clinical features ranges from severe childhood onset to late onset slowly progressive disease. Astejada et al., have described in a large series of EDMD patients with a mean age at onset being 10.1 and 3.3 years in pathogenic variants in EMD and LMNA respectively [14]. However, in the present study the median age at onset was much earlier (early 1st decade) in patients with variants in both EMD and LMNA genes. Generally joint contractures occur during the first decade followed by muscle weakness which is typically described in EDMD1. However, in EDMD2 contractures may appear after onset of muscle weakness [13]. The most common sites include elbow, ankle and posterior cervical muscles [3]. In the present study all patients with EMD had ankle contracture, while in LMNA, the ankle, elbow, knee and hip joints were equally involved. Neck contractures occurred in only one patient each with LMNA and EMD pathogenic variants. In a study done on LMNA related muscular dystrophies in Chinese cohort by Fan et al, contractures were noted in 75% of EDMD2 with ankle being the first joint involved followed by elbow joint [15]. Prominent finger contractures similar to collagenopathies was noted in two patients, one each with EMD and SYNE1 variants, which has not been reported previously. Serum CK level ranges from normal to moderately elevated [3]. A similar trend was noted in our patients.
Comparison with previous studies is summarized in Table 2.
Comparison with previous studies on EDMD
OTHER LAMINOPATHY RELATED MUSCULAR DYSTROPHIES
Muscle biopsy done in EDMD2, MDCL and LGMD1B showed most frequently the features of myopathy followed by focal perivascular infiltrates in all these three phenotypes. Studies by Komaki et al., [31] and Quijano-Roy et al., [29] have shown that inflammatory infiltrates were noted commonly in MDCL especially in the perimysial connective tissue. However, Fan et al., [15] have shown that 50% of MDCL and 33.3% of EDMD2 patients in their cohort showed inflammatory infiltrates in the entire muscle biopsy specimen including necrotic/non-necrotic fibres, endomysium and perivascular region of the perimysium. To ascertain the diagnostic importance in laminopathies related muscular dystrophies of this finding similar to other conditions such as facioscapulohumeral dystrophy and dysferlinopathy requires further dedicated studies. All pathogenic variants in both MDCL and LGMD1B are missense heterozygous variants except for one patient with LGMD1B.
CONCLUSION
This study on muscular dystrophies associated with nuclear envelopathies shows phenotype and genotype variability. The salient features include ophthalmoparesis and facial weakness in LMNA (in one patient each) and upper limb phenotype with novel variation in SYNE1.
The occurrence of finger contractures in EMD and SYNE1 is a novel finding which may aid in specific genetic diagnosis.
Footnotes
ACKNOWLEDGMENTS
None.
FUNDING
K.P. holds a Canadian Institutes of Health Research postdoctoral fellowship award under grant no. MFE-491707.
DATA AVAILABILITY
Data will be available on request from the corresponding author.