
Abstract
Background:
The slow channel syndrome is a rare hereditary disorder caused by a dominant gain-of-function variant in one of the subunits of the acetylcholine receptor at the neuromuscular junction. Patients typically experience axial, limb and particularly extensor finger muscle weakness.
Objective:
Age at diagnosis is variable and although the long-term prognosis is important for newly diagnosed patients, extensive follow-up studies are rare. We aim to provide answers and perspective for this patient group by presenting an elaborate description of the lifetime follow-up of two slow channel syndrome patients.
Methods:
We describe 40 years follow-up in two, genetically confirmed cases (CHRNA1; c.866G > T p.(Ser289Ile)(legacy Ser269Ile) and CHRNE; c.721C > T p.(Leu241Phe)(legacy Leu221Phe) variants).
Results:
We find that the disease course has a fluctuating pattern and is only mildly progressive. However, hormonal imbalances, (psychological) stress or excessive hot or cold environments are often aggravating factors. Quinidine and fluoxetine are helpful, but ephedrine and salbutamol may also improve symptoms.
Conclusion:
Slow channel syndrome is mildly progressive with a fluctuating pattern. The observations reported here provide a lifespan perspective and answers to the most pressing questions about prognosis and treatment options for newly diagnosed patients.
Keywords
INTRODUCTION
Congenital myasthenic syndromes (CMS) are a rare group of disorders affecting the neuromuscular junction (NMJ) [1–3]. The NMJ acetylcholine receptor (AChR) is a pentameric ligand-gated ion channel composed of four different subunits 2(α1), β1, δ, ɛ. Slow channel syndrome (SCS) is caused by many different variants (currently 26 known) in the genes encoding any of the four subunits of the AChR [4–8]. It usually presents with autosomal dominant inheritance, although recessive patterns and de novo variants have been described [9, 10]. The mutations alter the kinetics of the AChR resulting in prolonged opening times of the channel or increased reopenings, which lead to repetitive compound muscle action potentials (CMAP). The kinetic abnormality results in calcium overload, widening of the synaptic space, and destruction of the junctional folds [11, 12]. The most prominent symptoms are weakness, mostly in bulbar, respiratory, axial and finger extensor muscles, with ptosis, and fatigue but these trend towards a fluctuating pattern [13].
Due to the rarity of the disease, a delay in diagnosis is common and the long-term disease course has only sporadically been reported [5, 13–15]. As a result, the progression of the disease, long-term prognosis, and (pharmacological) treatment options are the most pressing questions asked by newly diagnosed patients. We were able to follow-up for 40 years on the first two European SCS patients extensively described in 1987 [15]. Here we established the genetic variants, confirming the diagnosis of SCS and offer a lifespan perspective on the SCS disease course and treatment options.
PATIENTS AND METHODS
Patients
We performed a retrospective follow-up study of the two patients described by Oosterhuis et al. [15] which were the first SCS patients reported in Europe. Both cases were female, currently aged 66 years (Case 1) and 61 years (Case 2). The genetic diagnoses were made (see below) and in each case, a medical history, complete neurological examination and retrospective records were summarized. Symptoms were then assessed for every decade in the patient’s medical charts and supplemented with data extracted from the patient interviews at the Radboud University Medical Centre.
This study was approved by the ethical committee of the Radboud University Medical Centre (2017–4022), and informed consent was obtained for the release of anonymized patient information.
Genetic analysis
Exome sequencing was performed as previously described [16]. In summary, capture of exons was done using an Agilent SureSelect Human All Exon 50 Mb Kit (Santa Clara, CA, USA). Sequencing was performed using an Illumina Hiseq4000 (San Diego, CA, USA). Read mapping and variant calling were done using or BWA and GATK, respectively. A filter for a ‘muscle disorders’ gene panel was applied. This panel consists of ∼300 genes. Variant data were filtered by frequency (< 5%dbSNP, < 1%in house database, gnomAD (PMID: 34373650)) nucleotide and amino acid conservation and exon/intronic position (–8 to +8 into the introns). Accession numbers NM_000079.3 (CHRNA1) and NM_000080.4 (CHRNE), and HGVS were used for nomenclature of variants.
RESULTS
Case series
Figures 1 and 2 and Tables 1 and 2 show details of the symptoms and course of disease in the two patients, including non-pharmacological treatments. Full descriptions of their early presentations up to the time of diagnosis are given by Oosterhuis et al. in 1987 [15].
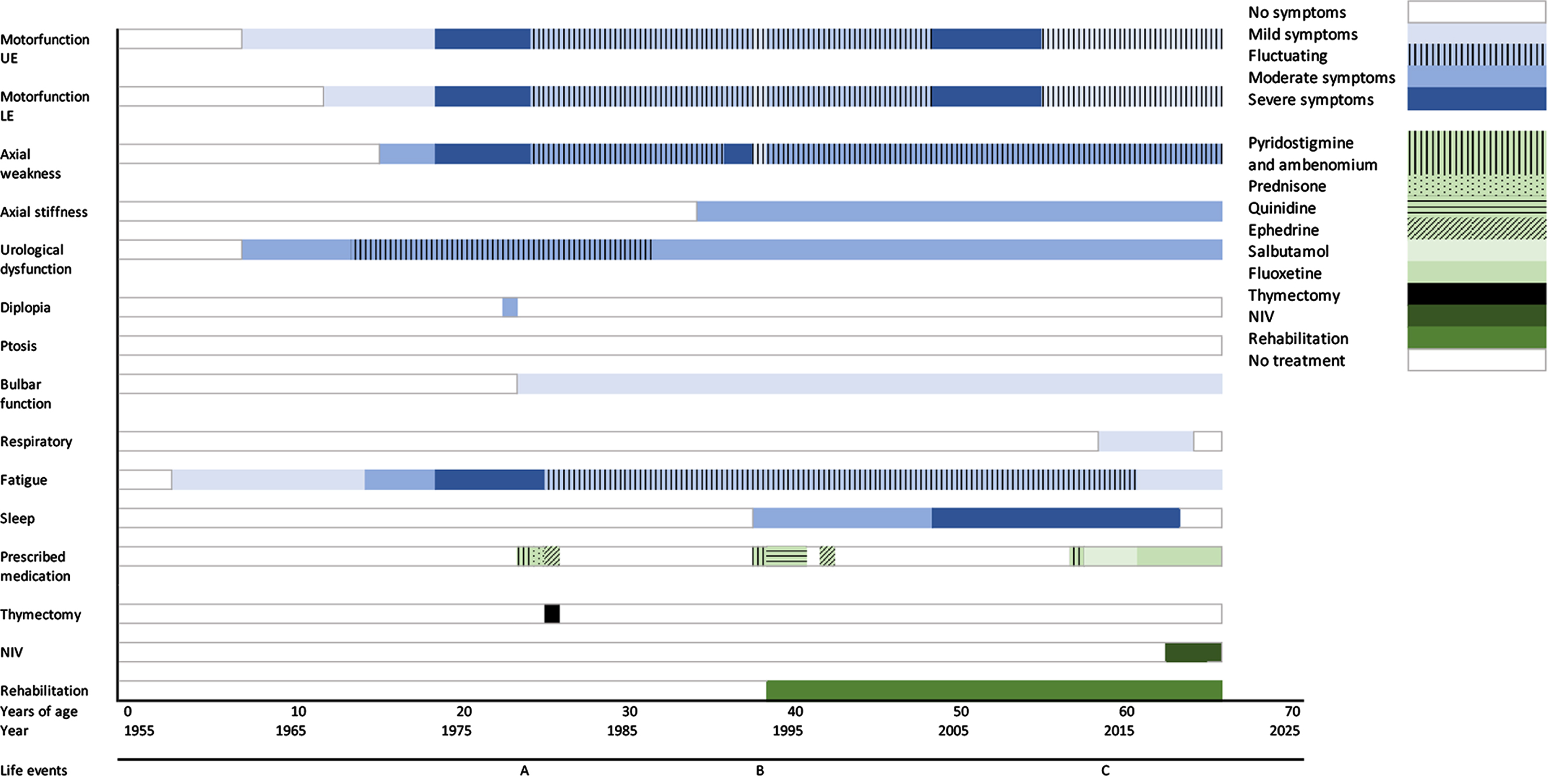
Overview of the course of disease including symptoms and treatments of case 1. UE = upper extremities, LE = lower extremities, NIV = non-invasive ventilation, SCS = slow channel syndrome. A = 1st child born, B = hysterectomy and adnexectomy, C = detection of SCS variant.
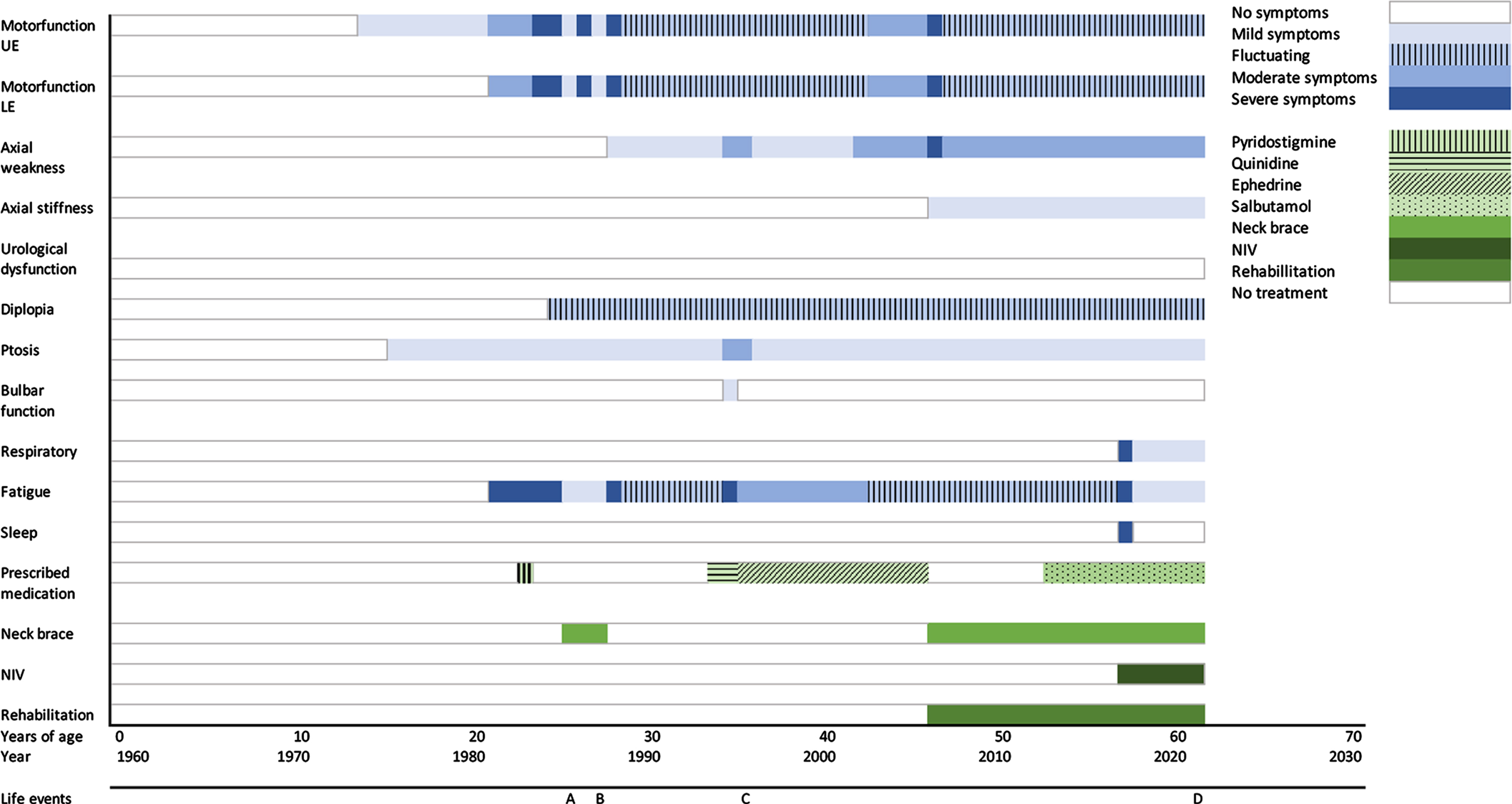
Overview of the course of disease including symptoms and treatments of case 2. UE = upper extremities, LE = lower extremities, NIV = non-invasive ventilation, SCS = slow channel syndrome. A = 1st child born, B = 2nd child born, C = premature menopause, D = detection of SCS variant.
Overview of the course of disease per decade including life events, treatments, and symptoms of case 1
Overview of the course of disease per decade including life events, treatments, and symptoms of case 2
Case 1
Her early years were characterized by weakness of the arms and persistent urinary incontinence. Leg weakness and choking episodes and some loss of fine motor skills were present by age 15 and deteriorated during menstruation. Further worsening was evident towards the end of her first pregnancy with increasing symptoms post-partum. At this time (1975), an EMG showed a decrement of 25%at 3 Hz stimulation, which made the neurologist suspect autoimmune myasthenia gravis (or possibly other neuromuscular transmission disorders), but she had an adverse reaction to pyridostigmine and ambenonium and no AChR antibodies were identified. However, since myasthenia gravis was still the most likely diagnosis at that time, thymectomy was performed but the thymus was pathologically normal. At age 25 her symptoms worsened further and, following the first reports in 1980 and 1982, a CMS was suspected. She was seen by a neurologist in a specialized tertiary clinic in London (UK) who demonstrated a repetitive response to a single nerve shock, similar to the reports from Engel et al. [2, 3] in 1979 and 1982, strong suggesting a CMS, specifically SCS.
Case 2
This patient did not record symptoms until her teenage years when she recalled ptosis, and weakness of facial and upper limb muscles. Finger extensor and leg muscle weakness were intermittent and by age 23, ptosis of 25%and lack of facial expression were reported by her treating neurologist. AChR antibodies were absent and pyridostigmine was deleterious. EMG showed a decrement at 3Hz stimulation and a repetitive muscle response to a single nerve stimulation. As in case 1, myasthenia gravis was thought possible but thymectomy was not scheduled because of doubt regarding the diagnosis. At age 26 her first son was born and two years later she gave birth to a second son. During two pregnancies in her 20s symptoms were mild but deteriorated quickly postpartum and only recovered slowly.
Subsequently symptoms worsened and quinidine was tried with only subjective and temporary improvement that was not sustained. With further deterioration she was prescribed ephedrine which appeared to stabilize symptoms initially but, following subsequent worsening symptoms, she stopped all medication. At this stage her fingers could no longer extend, and she had leg weakness. Like case 1, she reported deterioration of her muscle symptoms during excessive hot or cold weather.
From age 48 until 53, her symptoms had stabilized, and salbutamol started at age 53, produced good improvement of her symptoms. Similar to case 1, dysomnia and headaches were reported but shown to respond to nocturnal NIV. Currently cervical weakness and some weakness of arms and hand are present but manageable.
Oosterhuis et al. [15] reported that both the father and brother showed similar EMG repetitive responses at 0.2 Hz stimulation. Both father and brother reported no muscle weakness or other symptoms and had normal strength and endurance on examination at that time. Her father remained without symptoms throughout his life until his death due to pneumonia aged 63, and her brother did not develop symptoms until middle adulthood, after which they lost contact. Genetic tests have not been performed.
Genetic analysis
DISCUSSION
SCS is a rare condition which can now be diagnosed relatively easily by mutation analysis. Our patients developed symptoms after childhood, which were exacerbated by pregnancy or menstruation and showed a fluctuating pattern. These features make SCS prone to a misdiagnosis of autoimmune myasthenia gravis or a congenital myopathy. However, the long-term prognosis has not been described previously and in this retrospective follow-up study, we describe the course of disease and treatments of two cases with SCS.
In both patients the most prominent symptoms were neck and upper limb muscle weakness, impaired fine motor skills, proximal muscle weakness, ptosis and fatigue which has a fluctuating and mildly progressive pattern; these fluctuations are not only associated with hormonal imbalance during menstruation or post-partum, as previously reported [17], but also emotional or psychical stress and extremes of temperature as aggravating features. Dysomnia was another symptom experienced by both patients and appeared to be caused by nocturnal hypoventilation responding well to NIV.
These two women were the first SCS patients described from Europe and the gene mutations were not identified until subsequently reported in two case series, and diagnostically confirmed here [9, 18]. The c.866G > T; p.(Ser289Ile) variant in the CHRNA1gene and the variant c.721C > T;p.(Leu241Phe) in the CHRNE gene. Both mutations increase affinity for acetylcholine and slow channel closing consistent with the electrophysiological findings of Engel et al. [3] and Croxen et al. [14] which demonstrated a prolonged open time of the ACh-induced ion channel, the key feature of a slow channel syndrome [19, 20].
Treatment for SCS has evolved. Aggravation by drugs designed to increase acetylcholine is typical and improvement found with those that shorten channel open time. However, the effects can be slow to develop and ephedrine and salbutamol, which are helpful in patients with other forms of CMS in which AChR function is decreased, may also improve symptoms, as found here in case 2 [21, 22].
Due to the rarity of the disease and fluctuating pattern, diagnosis might be difficult and symptoms may be mistaken for seronegative myasthenia gravis [23]. However, SCS may be included in the differential diagnosis when a patient presents with cervical and upper limb weakness, shows no or adverse response to a cholinergic agonist such as pyridostigmine, no AChR or MUSK antibodies are detectable and EMG shows a repetitive CMAP evoked by a single nerve stimuli [24]. Although, one should keep in mind that repetitive CMAP can also be seen in other CMS due to end-plate AChR deficiency.
Electrophysiological studies play an important role in the assessment of patients with a suspected CMS and help differentiating between CMS and other neuromuscular disorders. This is generally much faster than genetic analysis thereby preventing unnecessary treatments. Furthermore, with the availability of next generation sequencing as a diagnostic tool, the diagnostic delay is expected to diminish.
Our study does have limitations. Firstly, recall bias causes a substantial challenge in a retrospective interview covering a period of 40 years, but we were able to validate answers from the very detailed medical records. Secondly, due to the rarity of the disease only two cases were included which may limit the generalizability to other SCS patients. Nevertheless, these observations may provide a lifespan perspective and answers to the most pressing questions about prognosis and treatment options for newly diagnosed patients with SCS.
Footnotes
ACKNOWLEDGMENTS
Several authors of this publication are members of the Radboudumc Center of Expertise for neuromuscular disorders (Radboud-NMD), Netherlands Neuromuscular Center (NL-NMD) and the European Reference Network for rare neuromuscular diseases (EURO-NMD).
FUNDING
No funding was received towards this work.
CONFLICT OF INTERESTS
The authors have no conflict of interest to report.