
Abstract
Congenital myasthenic syndromes (CMS) result from genetic mutations that cause aberrations in structure and/or function of proteins involved in neuromuscular transmission. The slow-channel CMS (SCCMS) is an autosomal dominant postsynaptic defect caused by mutations in genes encoding alpha, beta, delta, or epsilon subunits of the acetylcholine receptor resulting in a functional defect which is an increase of the opening time of the receptor. We report a case of SCCMS due to a heterozygous mutation in the M2 domain of the AChR alpha subunit - CHRNA1:ENST00000348749.6:exon7:c.806T>G:p.Val269Gly and corresponding kinetic defect. A substitution of valine with phenylalanine in the same position has been previously described. This is the first reported case of a new CHRNA1 variant in a patient with SCCMS from South Asia. We also highlight the phenotype that would favour a genetic basis over an autoimmune one, in an adult presenting with fatigable weakness.
Keywords
INTRODUCTION
Congenital myasthenic syndromes (CMS) result from mutations that cause aberrations in structure and/or function of proteins involved in neuromuscu-lar transmission. They comprise a group of genetically heterogeneous disorders that can be classified by the site of the transmission defect: presynaptic, synaptic, and postsynaptic. CMSs are rare and are frequently misdiagnosed or undiagnosed. Misdiagnosis was reported in 94% of adults with CMS with a median diagnostic delay of nearly 3 decades from symptom onset in a case series of 34 patients [1]. CMS can mimic several disorders and diagnosis may require specialized diagnostic methods. Due to whole exome sequencing (WES) targeting the entire set of human exons, increasing numbers of patients are diagnosed with CMS who have non-specific clinical symptoms or who have a diagnosis of congenital myopathy. The differentiation from autoimmune (acquired) myasthenia gravis is important, as CMS does not respond to either thymectomy or immunosuppressive therapy. Features that distinguish CMS from autoimmune myasthenia are: a positive family history, onset usually in childhood, absence of acetylcholine receptor (AChR) and/or MUSK antibodies and failed response to immune therapy [2, 3]. A negative family history however does not exclude it.
The slow-channel CMS (SCCMS) first described by Engel et al. in 1982, is an autosomal dominant disorder of the postsynaptic side of the neuromuscular junction [4] and may be caused by mutations in genes encoding alpha, beta, delta, or epsilon subunits of the AChR [4, 5]. The resultant functional defect is an increase of the opening time of the receptor, producing prolonged end-plate currents and excess calcium leakage into the end-plate region of the muscle fibre [4].
A single report of SCCMS due to the substitution of valine with phenylalanine in the AChR α-subunit at position 269 has been made previously [6]. Here we report a case of SCCMS with the missense mutation p.Val269Gly at this position and discuss clinical features that would favour a genetic basis over an autoimmune one, in an adult presenting with fatigable weakness.
CASE REPORT
A 42-year-old Sri Lankan man presented with subacute onset left index finger drop for 1 month. He reported a background history of fluctuating fatigability and weakness of proximal and distal upper limbs and proximal lower limb muscles accompanied by drooping of his eyelids for the preceding 14 years. There was no history of diplopia, bulbar symptoms, respiratory involvement and symptoms suggestive of facial and neck muscle weakness. He had been on pyridostigmine, prednisolone and azathioprine for 14 years with a clinical diagnosis of autoimmune myasthenia gravis seronegative for AChR antibodies. His symptoms had been fluctuating over this period with periods of exacerbations. However, over the last 2 years he had noted worsening of muscle fatigue despite medication. He specifically complained of intermittent wrist and finger drop during exacerbations. There was no family history of a neuromuscular disorder. There was no history of parental consanguinity. His birth history and early childhood development were unremarkable.
On examination, he had symmetrical partial ptosis with demonstrable fatigability and external ophthalmoplegia with limited eye movements in all directions. Rest of the cranial nerves were normal. He had distal asymmetrical (right > left) upper limb weakness predominantly involving the wrist and finger extensors (MRC grade 3/5). Deep tendon reflexes and the sensory examination were normal. The neck muscle power was normal and there was no evidence of respiratory muscle weakness or myotonia.
Specific antibodies (AChR, MuSK) for autoimmune myasthenia were negative (LRP4 antibodies seen in double seronegative patients were not tested due to unavailability of the test). Nerve conduction was normal, but electromyography (EMG) of the distal muscles (first dorsal interosseous and extensor digitorum communis) showed myopathic changes. Low frequency repetitive nerve stimulation (RNS) showed a marked decrement of more than 30%. Decremental response was defined as a > 10% decrease in amplitude or area of the fourth CMAP compared to the first CMAP on 2 Hz RNS of selected motor nerves (ulnar and accessory nerves).
A diagnosis of SCCMS was confirmed by genetic analysis and kinetic studies of the receptor (Oxford, United Kingdom). Informed consent to perform the above studies was obtained.
Mutation analysis
Next generation sequencing was carried out by Novogene (Hong Kong) using an Agilent SureSelect Human All Exon V6 kit to generate the library which was sequenced using an Illumina sequencer. A heterozygous variant was identified in CHRNA1 encoding the muscle AChR α-subunit and was confirmed by Sanger sequencing. The variant is CHRNA1:ENST00000348749.6:exon7:c.806T > G:p.Val269Gly. This transcript lacks the P3A exon and encodes the functional form of the muscle AChR α-subunit. In human muscle there are two alternatively spliced mRNA transcripts forms for the AChR α-subunit that generate two protein isoforms which are expressed at a 1 : 1 ratio, but the isoform that harbours the P3A exon is non-functional [7].
There were no other variants in known CMS genes identified.
Expression studies & Kinetic analysis
Expression of AChR was determined by detection of radiolabelled 125I-α-bungarotoxin bound to the surface of transiently transfected HEK293T cells (detailed experiment available at https://doi.org/10.1016/j.nmd.2013.10.009) [8]. Homozygous AChR harbouring the mutation p.Val269Gly had expression levels at 13.9 + /–0.8 % (n = 6) compared to wild-type AChR (WT). Cell-attached patch clamp recordings were made from transfected HEK293 cells [8]. Mutant channel activity was hard to find reflecting low cell surface expression of these channels. Only 5 out of > 12 patches from well transfected cells showed activity. When activity was found there were long periods (many tens of seconds) of no activity that were interspersed with clusters of activity. During 3106 seconds of recordings only 56 clusters were recorded, which included only 116 bursts of activity. Due to the low numbers of bursts recorded a full analysis is not possible, however, within these clusters of activity, bursts were extremely long, some more that 400 ms in duration. Of all bursts recorded 70% were longer than 10 ms and 45% were longer than 40 ms (typical long WT bursts are usually 5 to 20 ms in duration with a mean duration of ∼ 6ms (Fig. 1)).
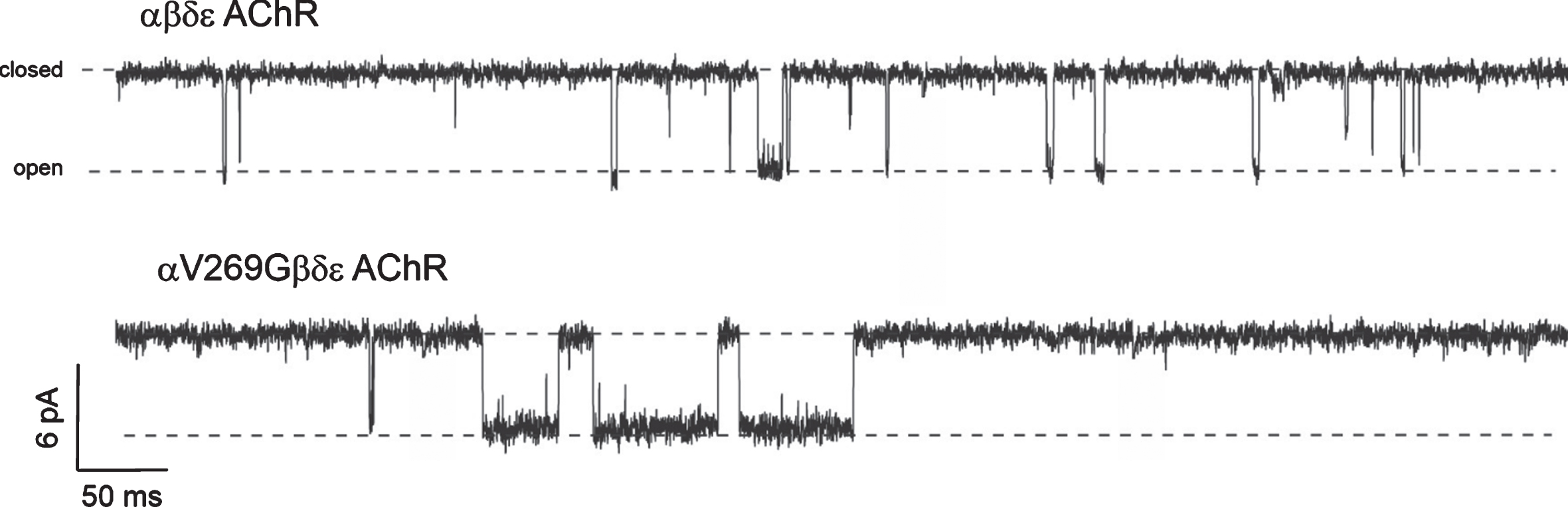
Example traces of recordings of activity with 100 nM ACh. Length of these example recordings is 800 ms. Upper trace shows wild type AChR channel activity, with regular channel openings (downward deflections), the longest bursts of activity between 5 and 10 ms. Lower trace shows example of prolonged bursts seen in recordings from mutant αV269G channels. The longest burst in this example was 68 ms.
These observations reflect two features of this mutation: (a) At low [ACh] of 100 nM or even 10 nM the channels were prone to desensitization (long periods of no channel openings). Normally desensitization is seen with WT channels at [ACh] at or above 10μM, (b) channel openings when they did occur as busts of activity were extremely prolonged. We were unable to define the extent of the burst duration prolongation since the periods of activity were so few, with long desensitized periods (thus burst duration histograms were not constructed). These two properties of channel activity are characteristic of SCCMS mutant AChR.
The patient was initiated on fluoxetine 10 mg/day and gradually increased to 80 mg/day. The immunosuppression and pyridostigmine were weaned off. He demonstrated improvement in wrist and finger extensors with near normal power at one-month. There was no change in the extent of ophthalmoparesis but ptosis was not evident. He remains clinically stable and independent in activities of daily living at 6 months of follow-up.
DISCUSSION
We report the first authenticated case of SCCMS of South Asian origin with a heterozygous mutation in the M2 domain of the AChR alpha subunit - CHRNA1:ENST00000348749.6:exon7:c.806T>G:p.Val269Gly and corresponding kinetic defect. A mutation at this residue, p.Val269Phe was first described in 1997 [6]. Here we identify a novel variant at this position, p.Val269Gly, that also generates a SCCMS, but with less severe clinical features.
Studies on the mutation conclude the following: a) there is reduced expression of the mutant AChR b) there is the presence of prolonged channel activation episodes and c) marked tendency of the mutant AChR to desensitize. This is similar to characteristics demonstrated by Milone et al. [6] thus supporting the phenotypic diagnosis of SCCMS. The lower expression of the mutant α-subunit may explain the relatively mild phenotype, suggesting that the majority of AChR channel openings at the endplate would have normal WT kinetics.
The clinical phenotype of SCCMS is variable [9]. In mild SCCMS phenotypes onset is usually after adolescence. SCCMS patients commonly present with severe weakness of the cervical, scapular, wrist, and finger extensor muscles [10]. However, in some, ptosis, ophthalmoparesis, dysarthria, dyspha-gia, proximal limb weakness, and respiratory insufficiency may occur [4, 10]. Our patient demonstrated predominant wrist and finger drop in addition to ophthalmoplegia with ptosis. Furthermore, his symptoms worsened with anti-cholinesterase medication as has been reported in SCCMS [11]. Despite not having a family history or a childhood onset, an SCCMS was clinically suspected in our patient because of the typical pattern of muscle involvement, the long history, poor response to immunosuppression, worsening with cholinergic medication and the marked decremental response on RNS even at 2 Hz. Our patient demonstrated an initial response to cholinergic medication. In many forms of CMS that involve pathogenic mechanisms that disrupt endplate structure there can be an initial positive response to cholinesterase inhibitors prior to the longer-term detrimental effect of these medications becoming apparent. We believe this is what occurred with our SCCMS patient.
Neurophysiology can be helpful in the diagnosis of SCCMS. A Repetitive Compound Muscle Action Potential (R-CMAP) after a single supra-maximal stimulus is often seen with SCCMS and is due to either excess acetylcholine (ACh) which remains after the first discharge or because a normal amount of ACh causes reactivation [12]. Congenital acetylcholinesterase (AChE) deficiency can also demonstrate similar electrophysiological features. However, this particular phenomenon of R-CMAP was not observed in our patient.
Short-duration, small-amplitude motor unit action potentials on EMG can be seen in NMJ disorders from intermittent blocking of some muscle fibre action potentials due to failed neuromuscular transmission [13]. This appearance was observed in the distal muscles of our patient and is often seen in myopathic disorders often leading to misdiagnosis or delay in diagnosis, emphasizing the importance of performing RNS. What can add to the confusion is that myopathies and neuromuscular junction disorders can co-exist, and may be observed in CMSs due to mutations in ALG2, ALG14, COL13A1, DOK7, DPAGT1, GMPPB or GFPT1 [14].
Fluoxetine and quinidine have been found to be of symptomatic benefit in treating SCCMS [15]. Both are long-lived open-channel blockers of the AChR with the capacity to block it in its open state and shorten the duration of otherwise prolonged synaptic currents [15]. Fluoxetine, which is thought to have a neuro-protective role and is used in doses up to 80 mg daily, was used with success at this level in our patient [15].
In conclusion, next-generation sequencing has facilitated the discovery of rare mutations in the diagnosis of CMS. Given that the same medication can be effective, ineffective, and even harmful depending on the mutation and its kinetic defect, our case report highlights the importance of accurate genetic diagnosis in CMS. It also helps to differentiate CMS from seronegative autoimmune myasthenia thus mitigating the requirement for immunosuppressive therapy and its long-term side effects. Furthermore, it helps to map the distribution of rare genetic mutations of CMS worldwide in different populations.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
FUNDING
None.
CONTRIBUTOR STATEMENT
IKG, SN and TC were involved with concept, drafting and editing of the manuscript. IKG, SN, KG and SS evaluated and managed the patient and edited the manuscript. LH extracted DNA from the patient and was involved in concept and editing of the manuscript. SM, RW, JC, DB were involved in genetic studies, kinetic studies and editing of the manuscript.