
Abstract
Mutations in PLEKHG5, a pleckstrin homology domain containing member of the GEF family, are associated with distal spinal muscular atrophy and intermediate Charcot-Marie-Tooth disease. Here, we describe an isolated case with distal intermediate neuropathy with scapular winging. By whole exome sequencing, we identified the homozygous PLEKHG5 Arg97Gln missense mutation, located in the N-terminal region of the protein. This mutation resides between a zinc-finger motif and a RBD domain, involved in binding rnd3, a RhoA effector protein. We conclude that based on the characteristic phenotype presented by the patient and the supportive genetic findings, the PLEKHG5 mutation is the causative variant.
Keywords
INTRODUCTION
Until recently, only six biallelic variants in PLEKHG5 had been linked with distal spinal muscular atrophy type 4 (DSMA4) and intermediate Charcot-Marie-Tooth (CMT) disease reported in five unrelated families (Fig. 1) [1–4]. A recent study reported nine additional families with most prominently proximal weakness, extending the spectrum of PLEKHG5-related disease (Fig. 1) [5]. Four other recent reports describe five additional patients with lower motor neuron disease or intermediate CMT with biallelic PLEKHG5 mutations (Fig. 1) [6–9].
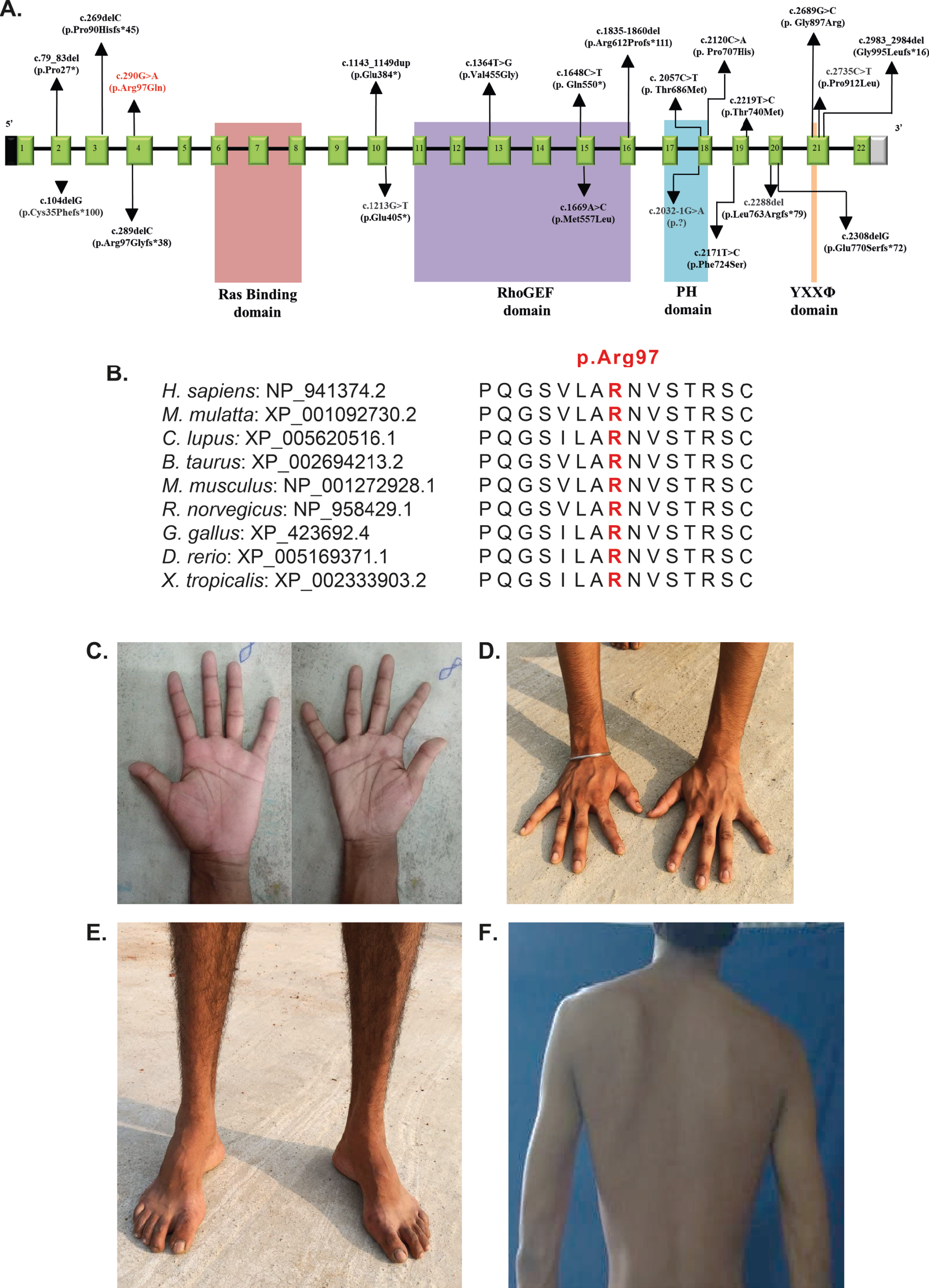
(A). Schematic representation of the variations identified in PLEKHG5 gene with corresponding exons and protein domains, Ras binding domain (RBD), RhoGEF/Dbl Homology (DH) domain, Plekstrin Holomology (PH) domain, and tyrosine-based sorting motif domain (YXXΦ). The 22 exons of PLEKHG5 (NM_198681.3/ENST00000377748.1) are represented as boxes with respective exon numbers with non-coding regions shaded in black and grey at the ends. The protein domains are depicted according to the Uniprot database and recent literature (6, 9). The novel variant found in our is study is marked in red. The size of exons /introns is not represented at scale. (B). Amino acid conservation at position 97 across species. (C, D) Note the atrophy of the small muscles of the hands and distal forearms. (E) Atrophy of the distal leg and foot muscles with pes cavus. (F) Scapular winging. Permission was granted by the patient to use these photos in publication.
PLEKHG5 encodes a pleckstrin homology do-main containing member of the GEF family, that is mainly expressed in the nervous system [10, 11]. Many PLEKHG5 mutation-specific effects are still unknown, but modelling of PLEKHG5-deficient systems has revealed several important key functions. PLEKHG5 has a prominent role in regulating autophagy of synaptic vesicles in motoneurons [12]. It performs this function by acting as a guanine exchange factor for Rab26, a small GTPase delivering synaptic vesicles to autophagosomes [13]. Reduced PLEKHG5 expression results in similarly reduced Rab26 activity, resulting in defective autophagy with subsequent accumulation of synaptic vesicles [12].
Here, we report one additional patient with the classic distal-onset intermediate CMT phenotype and a novel, homozygous PLEKHG5 missense mutation in the N-terminal region located between a putative zinc-finger motif and an RBD domain.
CASE REPORT
An 18-year-old male of Indian ancestry, born to healthy non-consanguineous parents, presented to the Neurology services in August 2015 with complaints of progressive lower limb weakness in the previous 18 months, altered gait, atrophy of limbs, distal more than proximal, as well as pes cavus and hammer toes. In the previous 8 months, he also noticed difficulties in running and walking fast. Additionally, he reported pressure-induced sensory symptoms in the extremities.
At the examination, the patient had slender habi-tus, demonstrated asymmetric (left more than right) scapular winging, bilateral atrophies of the supraspinatus, deltoid, intrinsic hand muscles, as wells as muscles of the anterior and lateral compartments of the lower legs and feet. He displayed a bilateral steppage gait and postural tremor in the fingers. On palpation, ulnar nerves were mildly thickened and distal hypotonia of the upper limbs was noticed. Manual examination of muscle strength demonstrated an overall distal muscle weakness pattern from the following MRC grading: 5/5 in the shoulder girdle, elbow, wrist, finger flexors and extensors, pelvic girdle, knee and ankle and toe plantar flexors, ankle invertor group of muscles. Ankle eversion was 2/5, ankle and toe dorsiflexors showed grade 0/5 strength only. The small muscles of the hands were 3/5 and the small muscles of the feet 1-2/5, all graded by the modified MRC scale.
There was 25 to 30%impairment in pinprick sensation in the hands and feet as well as mild reduction of vibration perception at medial malleoli. Tendon reflexes were absent in all four limbs (Table 1). Nerve conduction studies showed reduced compound muscle action potentials (CMAP) and absent sensory nerve action potentials (SNAP) as well as reduced motor nerve conduction velocities, corresponding to intermediate sensorimotor neuropathy, showing a combination of demyelinating signs and axonal loss (Table 1). His serum creatine kinase level was normal at 168U/L. Pure tone audiometry showed a mild unilateral loss in low frequency conduction, unrelated to the neurodegenerative phenotype. Overall, the phenotype corresponded to an intermediate sensorimotor neuropathy with scapular winging.
Nerve conduction studies of the patient
APB: Abductor pollicis brevis, ADM: Abductor digiti minimi, CMAP: compound muscle action potential, EDB: extensor digitorum brevis, MNCV: motor nerve conduction velocity, SNAP: sensory nerve action potential, SNCV: sensory nerve action potential, TA: tibialis anterior. Values out of the normal range indicated in bold.
Whole exome sequencing in this patient detected a likely pathogenic homozygous PLEKHG5 missense variant NM_198681.3: c.290G > A, p.Arg97Gln (Fig. 1). Sanger sequencing verified the heterozygous state in the non-consanguineous parents. This variant has overall strong predictions of pathogenicity: CADD 16.15; GERP (RS score) 4.44; MutationTaster Disease causing; PolyPhen Probably damaging; PROVEAN (Protein Variation Effect Analyzer) Tolerated; VEST3 0.43, SIFT Damaging. The Arg97Gln mutation affects a highly conserved residue and is only present once in heterozygous state in the reference genome database GnomADV2.1.1 and absent from the GenomeAsia 100k database [14, 15]. We identified no candidate mutations in other known neuromuscular disease genes fitting with the inheritance pattern.
DISCUSSION
The patient reported here, is of Indian ancestry from the West Bengal region and carries a homozygous PLEKHG5 missense variant Arg97Gln located in the N-terminal region of the protein. The distal sensorimotor neuropathy phenotype with scapular winging observed in this patient is very reminiscent of the initial PLEKHG5 phenotype reported [1, 2]. The combination of the very consistent phenotype and the genetic strength of the variant make the homozygous PLEKHG5 Arg97Gln missense variant the most likely genetic cause in this patient. Scapular winging is a recognizable and specific feature in this patient, as few genetic neuropathy subtypes besides PLEKHG5-neuropathy are associated with this symptom, only occurring in some patients carrying TRPV4, BICD2 in rare cases HSPB8 mutations [16–19]. These are genes also associated with (distal) myopathy and spinal muscular atrophy, in which scapular winging is a more common symptom.
Previously reported mutations in PLEKHG5 have been either truncating or missense variants (Fig. 1). The truncating variants are located throughout the protein, but the reported missense mutations seem to specifically cluster in the RhoGEF/Dbl homology (DH), Plekstrin homology (PH), and tyrosine-based sorting motif YXXΦ domains of PLEKHG5. These domains are involved in guanine nucleotide exchange, activation of NF-κB signalling pathway, and sorting of transmembrane proteins, respectively. The mutation we report here is different in the regard that this missense variant is located in the N-terminal region of the protein. While not located in a specific known domain or motif, there is a putative zinc-finger motif upstream and the Ras binding domain (RBD) domain downstream of the Arg97 residue. The physiological function of the arginine residue at position 97 is not yet known, but the location near the RBD binding domain suggests that this arginine residue could be important for binding to rnd3, a RhoA effector [20]. Subsequently, due to changes in PLEKHG5 binding with rnd3, a direct change in RhoA activation could occur. This would not be a first, as aberrant RhoA activation is also known for neuropathy-associated TRPV4 mutations and diabetic neuropathy [21, 22]. However, another mutational mechanism such as protein instability resulting in loss of protein expression cannot be excluded, thereby fitting with known truncating mutations in PLEKHG5. Interestingly, the c.290G > A, p.Arg97Gln mutation could also potentially result in loss of function via activation of a cryptic splice acceptor site, potentially resulting in altered splicing as predicted by Human Splice Finder [23]. All these remain potential explanations until further evidence is presented.
In conclusion, we describe a patient with PLEKHG5-associated intermediate Charcot-Marie-Tooth disease, with a novel homozygous Arg97Gln missense mutation. This case is particularly interesting due to the patient’s Indian ancestry, which is generally an underreported population, as well as the unusual location of the Arg97Gln mutation in the N-terminal region of the protein.
ETHICAL APPROVAL
Ethical approval for this study was obtained from the NIMHANS Ethics Committee for protocol title “Charcot Marie Tooth Disease Type 4: A deep phenotyping and functional analysis”.
CONFLICTS OF INTEREST
The authors have no conflicts of interest to report.