
Abstract
Background:
In many developing regions, genetic data on Charcot–Marie–Tooth disease (CMT) remains scarce.
Objective:
This study aimed to investigate the genetic landscape of CMT in Vietnam to guide the development of cost-effective diagnostic algorithms for patients with suspected genetic neuropathies.
Methods:
We recruited 44 patients with a diagnosis of CMT from three tertiary centers between March 2021 and December 2023 and recorded their clinical and electrophysiological characteristics. All patients were analyzed for duplications or deletions of PMP22, GJB1, MPZ, and MFN2 via multiplex ligation-dependent probe amplification (MLPA) and for 94 genes via targeted next-generation sequencing (NGS). The identified variants were classified per the American College of Medical Genetics and Genomics 2015 guidelines using VarSome, a bioinformatics engine.
Results:
Among 44 patients, 24 carried a total of 26 variants. Of these 26 variants, 15 were (57.7%) pathogenic, 6 (23.1%) were likely pathogenic, and 5 (19.2%) were variants of uncertain significance (VUS). Excluding the VUS, the diagnostic yield of the targeted sequencing was 43.2% (19/44). Through MLPA, PMP22 duplications were identified in 10 patients with the demyelinating type of CMT and 1 patient with the unclassified CMT type. The combined yield of MLPA and gene panels was 68.2% (30/44). We detected three novel pathogenic/likely pathogenic variants in GJB1, INF2, and IGHMBP2, as well as three novel VUS in MPZ, PMP22, and INF2. IGHMBP2 may represent the most prevalent autosomal recessive gene associated with CMT in Vietnam.
Conclusions:
We propose a sequential genetic testing approach for CMT in resource-limited settings, with the initial testing via MLPA for demyelinating CMT, followed by NGS for those who test negative. Our findings broaden the CMT genotype–phenotype profile of the Vietnamese population by identifying six novel candidate variants.
Keywords
Introduction
Charcot–Marie–Tooth disease (CMT) is the most common inherited peripheral neuropathy. With the development of advanced molecular techniques, its genetic heterogeneity has become increasingly evident – more than 90 causative genes have been identified to date.1–4 Despite variations in genomic profile across distinct ethnic populations, PMP22 duplication, resulting from a 1.5-Mb tandem duplication in chromosome 17p11.2–p12, remains the most common causative mutation, accounting for up to 50% of demyelinating CMT cases. 5 To investigate such a large duplication, gene multiplex ligation-dependent probe amplification (MLPA) is the gold-standard technique, preferred over approaches such as chromosomal microarray. Hence, MLPA is routinely performed as the first genetic investigation in patients who present with a significant reduction in motor nerve conduction velocity (MNCV) (≤38 m/s), a hallmark of demyelinating CMT. 6 An alternative strategy involves using a sequential molecular diagnostic algorithm with a more detailed MNCV classification combined with the patient's age at onset. 7 This approach involves testing one candidate gene at a time using Sanger sequencing, moving to the next most likely gene if results are negative. While systematic, this step-wise approach is time-consuming and may fail to detect rare CMT variants, reducing its practicality in the era of high-throughput NGS techniques. In the last five years, advanced pipelines for NGS analysis have exhibited superior efficiency to traditional methods in detecting single nucleotide variants (SNVs) and copy number variations (CNVs), offering higher sensitivity and faster turnaround times at a lower cost for an unlimited number of genes.8–11 Despite these advantages, implementation of NGS in resource-limited settings faces significant challenges due to costs, infrastructure requirements, and supply chain constraints.
The genetics of CMT in Vietnam remains largely unexplored. Therefore, we aimed to elucidate the genotype–phenotype relationships of CMT and to assess the yield and effectiveness of MLPA and NGS-based gene panels. Our findings are expected to contribute to the development of an algorithmic, effective, and accurate approach for the genetic diagnosis of CMT.
Materials and methods
Patient selection and data collection
We recruited patients newly diagnosed with CMT who had not yet undergone genetic testing from three tertiary centers in Vietnam, namely, Military Hospital 175, University Medical Center of Ho Chi Minh City (UMP), and Children's Hospital 2. Consistent with standard clinical practice, patients were defined as having CMT if they presented with symmetric, chronic, deforming, and length-dependent motor and sensory deficits, with or without a family history. These patients were identified after ruling out other causes of chronic neuropathies, such as chronic inflammatory demyelinating polyradiculoneuropathy. The CMT diagnoses were confirmed by a consensus panel of neuromuscular specialists. Nerve conduction studies (NCS) and electromyography (EMG) were performed to support the diagnosis, particularly in suspected cases of CMT type 1 (CMT1), which is typically characterized by diffuse slowing of nerve conduction velocities. 12 Patients suspected of having other genetic neuropathies (e.g., hereditary sensory or motor neuropathies), and multisystemic diseases, such as Friedreich's ataxia, Refsum disease, or mitochondrial neuropathies, were excluded. We also excluded patients with incomplete clinical, electrophysiological, and genetic data.
Genetic sequencing
Chemical reagents used in this study were NextSeq 2000 Reagents Kit (Illumina, USA). Genomic DNA extraction was performed using the MagMAX™ DNA Multi-Sample Ultra 2.0 Kit (Thermo Fisher Scientific, USA). Library preparation was conducted with the NEBNext® Ultra™ II FS DNA Library Prep Kit for Illumina (New England Biolabs, USA). xGen® Lockdown® Reagents (IDT DNA, USA) were used to enrich DNA fragments in the target gene region. Sequencing was carried out on a NextSeq (Illumina) with an average coverage of approximately 100×, ensuring that at least 95% of the target gene region had coverage above 10×. The sequencing data were aligned to the GRCh38 reference genome to identify genetic variants. Variants with a population frequency below 1%—as determined by existing Vietnamese genetic databases, including the 1000 Exome Sequencing Project and the Exome Sequencing Project (ESP)—were further classified using the ClinVar database, which is maintained by the US National Institutes of Health, at the time of reporting. The classification system comprised three groups: (1) pathogenic and likely pathogenic (P/LP), (2) variants of uncertain significance (VUS), and (3) benign and likely benign. Only pathogenic variants potentially associated with the patients’ clinical features were included in the final results. Laboratory validation indicated that the test's sensitivity and specificity exceeded 99%. The surveyed genetic variants included point mutations, as well as small deletions and insertions (<10 nucleotides) located in the coding and intronic regions (±10 nucleotides from an exon). The test had limitations in detecting variants outside the coding region, large deletions and insertions (>100 nucleotides), continuous short repeats, CG-rich regions, highly homologous sequences (e.g., pseudogenes), and mosaicism.
All patients underwent multiplex ligation-dependent probe amplification (MLPA) using two probe sets to detect duplications or deletions in PMP22/GJB1 and MPZ/MFN2, analyzed with Coffalyser software. Concurrently, they were tested using a basic CMT gene panel comprising 80 genes on the MiniSeq (Illumina) platform. Subsequently, only patients with negative findings from the basic panel underwent analysis using an extended panel of 14 additional genes (Appendix 1). This dual-testing approach was adopted to minimize sample loss by reducing the need for multiple visits, especially for those residing far from the study sites. Additionally, because MLPA can produce false-positive results, the combined strategy ensured that no other variants responsible for the patients’ phenotypes were overlooked.
The targeted capture region included all coding exons and adjacent intronic sequences extending up to 20 base pairs upstream and downstream of each exon–intron junction. True-positive SNVs and short insertion–deletion variations (indels) were confirmed via Sanger sequencing using the 3500 Genetic Analyzer (Applied Biosystems). All variants were classified according to the 2015 American College of Medical Genetics and Genomics (ACMG) guidelines, utilizing VarSome Premium (version 11.11.0; South Asian ethnicity) and ClinVar (February 2024).13,14 The PM2 criterion was validated using Vietnamese genetic databases.15,16 To increase the precision of variant classification, we performed Sanger testing on the probands’ family members, with their informed consent, to confirm the presence of the same variants.
Clinical and neurophysiologic investigation
Three neuromuscular specialists performed clinical and neurophysiological assessments. They evaluated six major muscle groups, namely, shoulder abduction, elbow flexion, wrist extension, hip flexion, knee extension, and ankle dorsiflexion, to determine the total Medical Research Council (MRC) scores. An MRC score of ≤3/5 indicated moderate-to-severe weakness. A loss of reflexes out of proportion to the degree of weakness was identified as areflexia in the proximal upper limbs while muscle strength remained intact.
We performed motor conduction studies on the median, ulnar, tibial, and peroneal nerves, as well as antidromic sensory conduction studies on the median, ulnar, sural, and superficial peroneal nerves. The median and ulnar nerve MNCV threshold of 38 m/s was used to differentiate between demyelinating and axonal CMT7,12,17,18 (Table 1). If the MNCV exceeded 38 m/s, a demyelinating condition was ruled out. 6 Furthermore, if the nerves were unexcitable, defined by amplitudes <0.5 mV, the case was labeled as unclassified CMT.
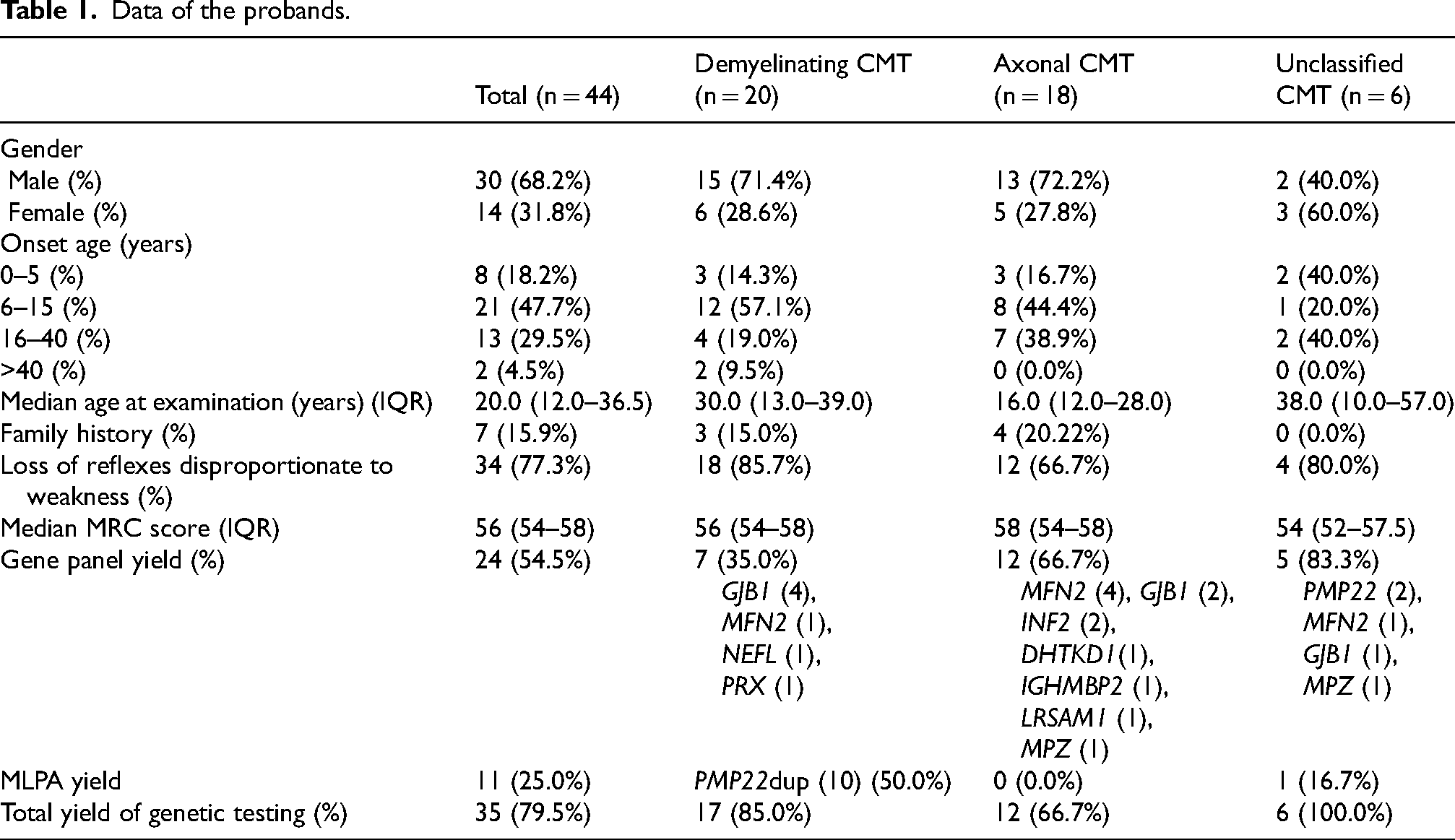
Data of the probands.
Statistical analysis
All statistical analyses were conducted using R software. Continuous variables with a normal distribution were presented as means and standard deviations (SD), whereas non-normally distributed continuous variables were reported as medians and interquartile ranges (IQR). For categorical variables, the Mantel–Haenszel chi-squared test was used for ordinal variables, and either the chi-squared or Fisher's exact test was applied for nominal variables. To compare non-normally distributed continuous variables between two independent groups, we employed the Mann–Whitney U test with exact distribution. Additionally, we applied a bootstrapping approach to calculate the 95% confidence interval (CI) for parameters without a normal distribution. A two-tailed P-value of <0.05 was considered statistically significant.
Ethical approval
Ethical approval was obtained from the Ethics Committee of the UMP (IRB-VN01002/IRB00010293/FWA00023448) on March 10th, 2021. All patients aged ≥16 years, or the legal guardians of the minors provided written informed consent prior to enrollment in the study. All research procedures adhered to ethical guidelines and were conducted in accordance with regulations of each participating hospital. None of the investigators reported any conflicts of interest.
Results
Demographic data
From an approximate total of 300,000 neurology outpatients, we enrolled 53 patients who met the inclusion criteria. Their ages ranged from 5 to 60 years (median: 20; IQR: 13–38), and 18 (34.0%) of them were female. Among these patients, 16 were members of seven unrelated families, leaving 44 index cases (Table 1). None of the families had consanguineous marriages. For clarity, all reported results refer exclusively to the index cases unless otherwise stated, and no enrolled cases were excluded.
Clinical and electrophysiological findings
Table 1 presented the findings on reflex loss disproportionate to weakness and the median MRC scores of 44 index cases. The MRC scores did not differ significantly between the axonal and demyelinating CMT groups (Mann–Whitney U test: W = 142, P = 0.25; median difference: −2, 95% CI: −3 to 2). Although a higher proportion of patients in the demyelinating CMT group exhibited areflexia disproportionate to weakness compared with the axonal CMT group (85.7% vs. 66.7%), Fisher's exact test revealed that this difference was not statistically significant (odds ratio = 0.36, 95% CI = 0.1–2.1, P = 0.26). Moreover, no patients presented with moderate-to-severe weakness (MRC scores ≤ 3/5) in the absence of muscle atrophy.
Table 3 presented a comparison of the characteristics of patients with and without PMP22 duplications within the demyelinating CMT subgroup. Patients harboring PMP22 duplications tended to have lower MRC scores and sought medical care at a younger age, with 50% presenting before the age of 13. Contrarily, 75% of the patients with other demyelinating types sought care after the age of 26 and had higher MRC scores. However, despite their seemingly better functional status, patients without PMP22 duplications exhibited significantly lower MNCV (Figure 1). The 97.5th percentile for MNCV of the group with PMP22 duplications was 23.0 m/s (95% CI: 19.3–23.4 m/s).
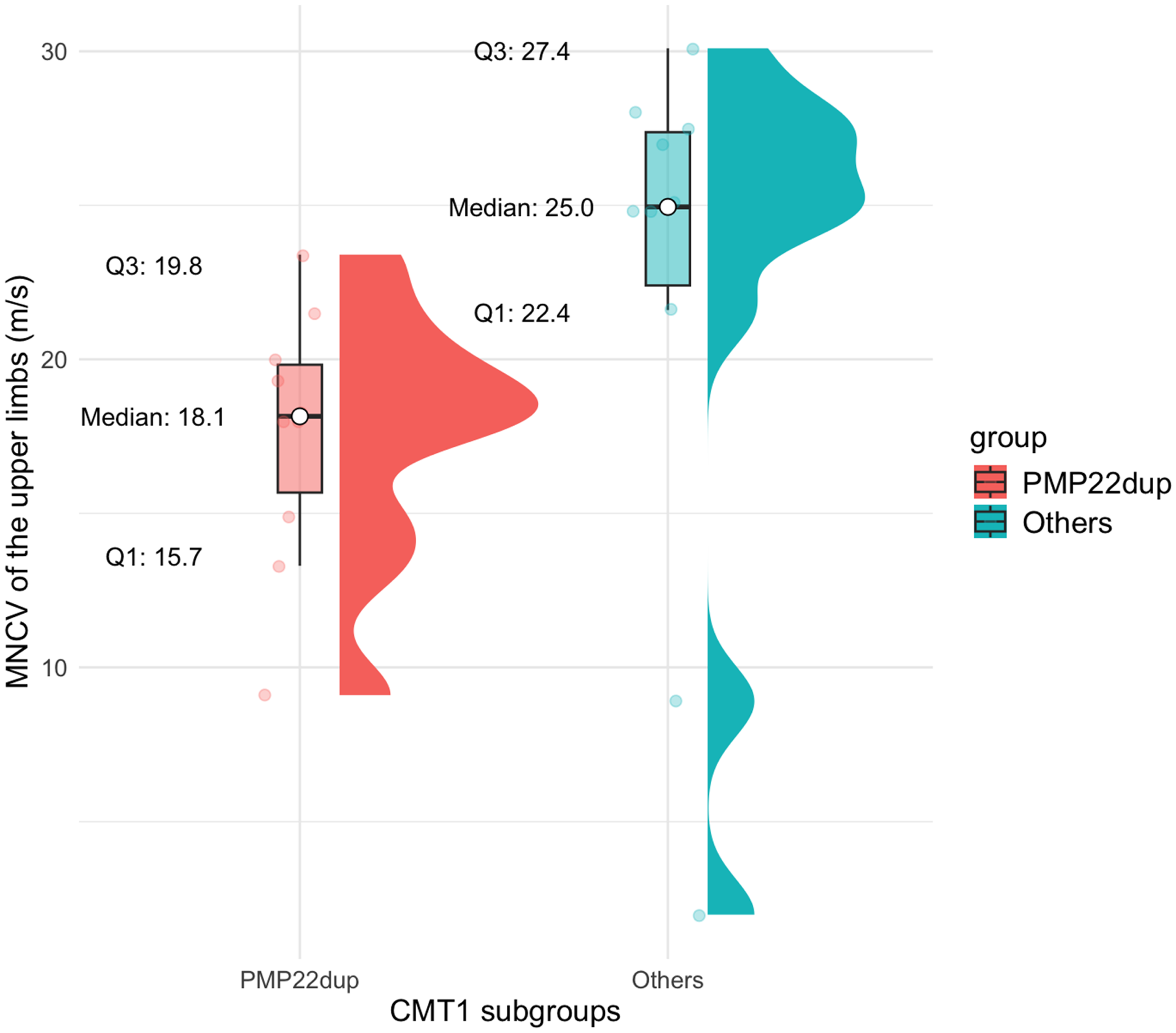
Comparison of upper limb MNCV for PMP22 duplication and nonduplication groups.
Genetic reports
The genetic results of the 44 probands are presented in Tables 2 and 3. Other family members not explicitly mentioned also demonstrated co-segregation of the variant. A total of 31 patients, including 11 patients from five distinct families, were identified via targeted NGS, with 33 variants. All patients within the same family lineage shared identical variants. When only index cases were considered, 24 of 44 patients harbored 26 unique candidate variants. Of these 26 variants, 15 (57.7%) were classified as pathogenic, 6 (23.1%) as likely pathogenic, and 5 as (19.2%) VUS. Excluding the VUS, the diagnostic yield of the targeted sequencing was 43.2% (19/44), and the combined yield of MLPA and gene panels was 68.2% (30/44). Among all the candidate variants, six were novel. These included three pathogenic/likely pathogenic variants: NM_000166(GJB1):c.284T > C (pathogenic), NM_001031714(INF2):c.2595C > A (likely pathogenic), and NM_002180(IGHMBP2):c.1132G > A (likely pathogenic), and three VUS: NM_000530(MPZ):c.553C > G, NM_000304(PMP22):c.335_337dup, and NM_001031714(INF2):c.454A > G (Table 2, Appendix 2). Autosomal recessive carriers of PRX and IGHMBP2 accounted for 6.5% (2/31) of the mutation-confirmed cases.
Summary of variants and clinical features in CMT index cohort.
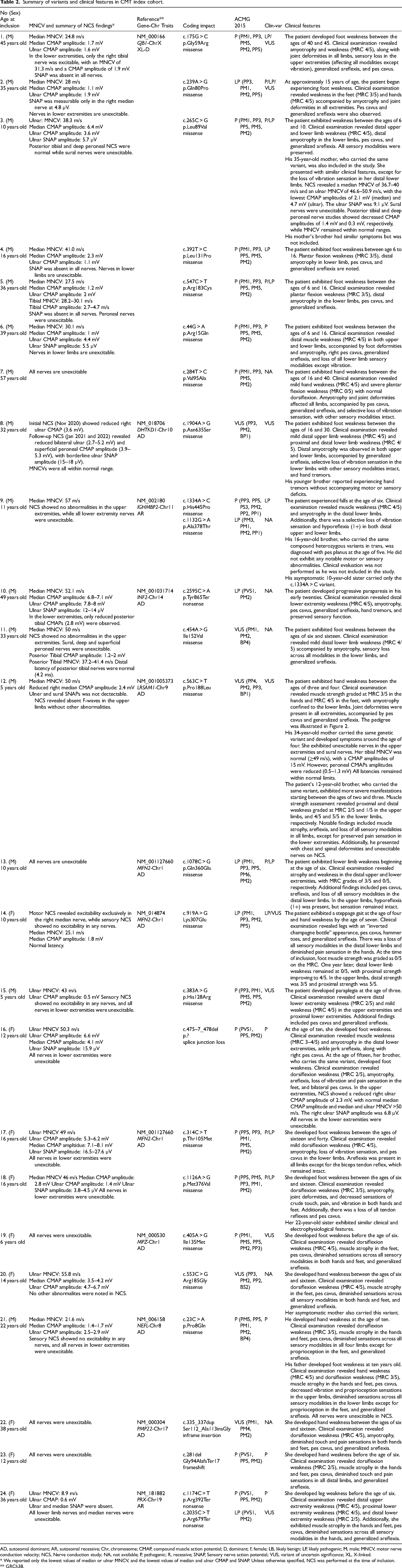
AD, autosomal dominant; AR, autosomal recessive; Chr, chromosome; CMAP, compound muscle action potential; D, dominant; F, female; LB, likely benign; LP, likely pathogenic; M, male; MNCV, motor nerve conduction velocity; NCS, Nerve conduction study; NA, not available; P, pathogenic; R, recessive; SNAP, Sensory nerve action potential; VUS, variant of uncertain significance; XL, X-linked.
* We reported only the lowest values of median or ulnar MNCV, and the lowest values of median and ulnar CMAP and SNAP. Unless otherwise specified, NCS was performed at the time of inclusion.
** GRCh38.
Comparison between patients with PMP22 duplications and other demyelinating CMT cases.
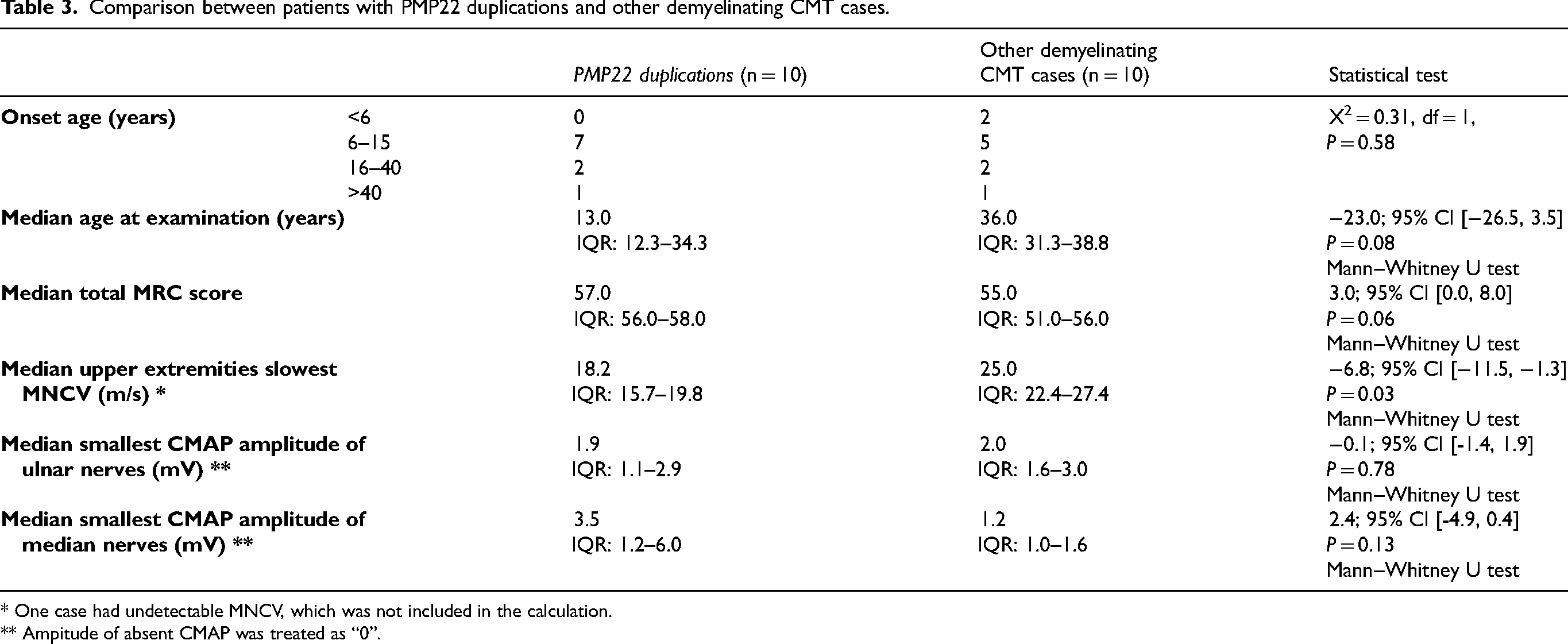
* One case had undetectable MNCV, which was not included in the calculation.
** Ampitude of absent CMAP was treated as “0”.
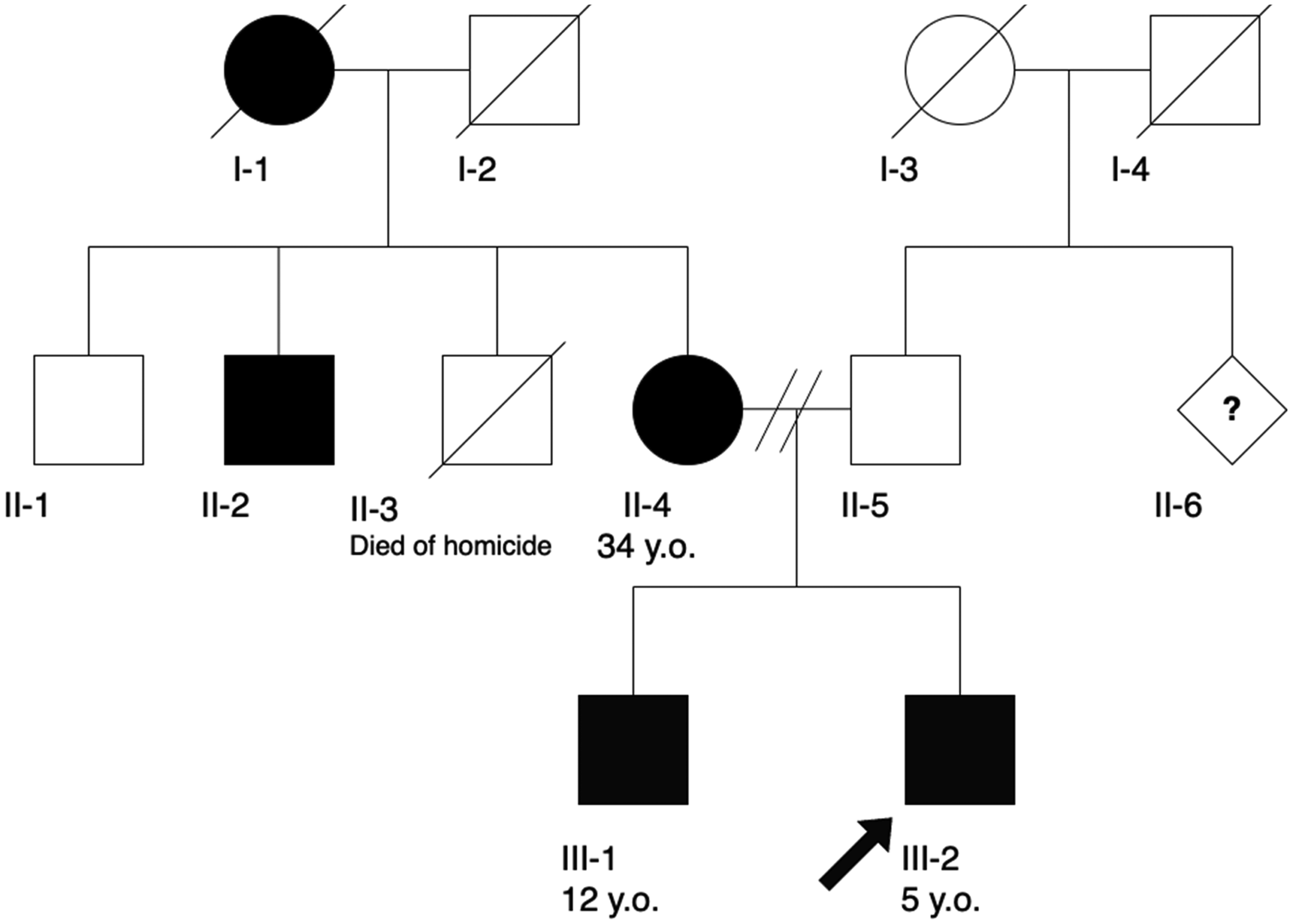
Pedigree of the family carrying LRSAM1:c.563C > T.
For axonal CMT cases, MLPA did not detect any duplications or deletions in PMP22, GJB1, MPZ, and MFN2. However, MLPA revealed PMP22 duplications in 10 patients with demyelinating CMT and in 1 patient with an unclassified CMT subtype. Additionally, NGS identified SNVs in PMP22 in two other patients, resulting in an overall PMP22 mutation prevalence of 37.1% (13/35) among the genetically confirmed cases. After PMP22, GJB1 and MFN2 were the second and third most prevalent genes, respectively, accounting for 20.0% (7/35) and 17.1% (6/35) of positive cases, respectively. When combined with MPZ (2/35) and INF2 (2/35), these five genes accounted for 85.7% (30/35) of all cases with positive genetic findings (Table 1).
Discussion
The male-to-female ratio in our study was approximately 2:1, a demographic distribution that closely mirrors the findings of the only other published study on CMT in Vietnam, which reported a male-to-female ratio of approximately 1.6:1. 19 Such a disparity may be attributed to health inequalities that systematically limit women's access to specialized healthcare services. Women, particularly those belonging to minority ethnic groups or residing in rural regions, may have less access to tertiary medical centers than men. Moreover, the chronic underservicing of neurology in Vietnam's healthcare infrastructure—characterized by a shortage of neurologists in primary and secondary healthcare facilities—further exacerbates patient referral challenges. Consequently, these compounded socioeconomic barriers may result in the substantial underrepresentation of women, particularly those from marginalized backgrounds, in our study.
This is the largest study in Vietnam to investigate the genetic spectrum of CMT using MLPA and NGS-based gene panels. The combination of these tests yielded an overall mutation detection rate of 68.2% (30/44), which aligns closely with the rate reported in a Taiwanese study (73.1%). 3 The PMP22, GJB1, MFN2, and MPZ genes consistently accounted for approximately 90% of genetically confirmed cases, regardless of ethnic populations.2–5,20 However, the emergence of INF2 (5.9%) in the Vietnamese population warrants confirmation in larger studies.
The prevalence of PMP22 mutations in our probands was 37.1%, a proportion substantially lower than the 69.2% previously reported in another Vietnamese cohort. 19 This discrepancy can be attributed to two key demographic factors: the median age and the age of disease onset. Our cohort exhibited a notably older median age compared to the reference group (20 vs. 13.5 years), and a lower proportion of early-onset cases (18.2% vs. 32.3% of patients experienced onset before the age of six) (personal communication, Trung Hieu, MD). 19
Although some authors have advocated PMP22 duplication/deletion analysis as the initial molecular diagnostic test irrespective of phenotype, the adoption of this practice can pose significant challenges in middle- to low-income countries. 9 For instance, in Vietnam, MLPA testing is confined to three facilities with limited capacity, and its availability is further constrained by frequent supply chain disruptions. Moreover, our study and that of Trung Hieu et al. have demonstrated the ineffectiveness of MLPA in the axonal subgroup. 19 These findings should dissuade clinicians from routinely requesting MLPA without prior electrophysiological assessment. Whenever the MNCV exceeds 38 m/s, NGS should be considered as an appropriate first-line test for a definitive diagnosis. 21 Similarly, analyzing GJB1, MPZ, and MFN2 for duplications or deletions is not warranted until PMP22 duplications have been ruled out and NGS has not identified any mutations. As expected, deletion of PMP22 was not observed in our cohort as it is associated with hereditary neuropathy with liability to pressure palsies, a distinct entity from CMT. A rare exception is that CMT1A may result from a compound heterozygous recessive SNV of PMP22 and a 1.5-Mb deletion in 17p11.2–p12. 22
A disproportion between reflexes and weakness is often considered a clinical hallmark of myelinating disorders. 23 Although the extent of weakness and the presence of reflex abnormalities can vary significantly depending on the specific CMT subtype and underlying genetic mutation, 24 the clinical features of our cohort were relatively uniform. Consequently, these characteristics were not useful for distinguishing between demyelinating and axonal CMT (Table 1). This uniformity could be attributed to the chronic rather than acute nature of the disease, which leads to secondary axonal damage and results in mixed clinical features. To our knowledge, no previous studies have investigated this matter.
Numerous studies, including ours, have documented significantly lower MNCV values in patients with PMP22 duplications compared to those with other types of demyelinating CMT (Figure 1, Table 3).3,7 In the PMP22 duplication group, the 97.5th percentile for MNCV was 23.0 m/s (95% CI: 19.3–23.4 m/s). This finding suggested that the categorization of the intermediate CMT subtype (25–45 m/s) does not influence the genetic testing approach, as all patients with demyelinating CMT (<38 m/s) have undergone MLPA, leaving no instances of PMP22 duplication within the remaining MNCV range of 38–45 m/s.3,12 However, the intermediate CMT classification remains valuable for establishing genotype–phenotype correlations.
Other factors–including age at onset, age at examination, total MRC score, and median and ulnar CMAP amplitudes–did not exhibit statistically significant differences between patients with and without PMP22 duplications. However, the difference in total MRC scores might be noteworthy, with a 95% CI of 0.0–8.0. This subtle difference in total MRC scores may reflect a relatively milder dysfunction in CMT1A, potentially due to the broader phenotypic spectrum observed in patients with demyelinating CMT associated with other genetic mutations, such as GJB1. 25 Besides, while the clinical dysfunction in both groups appeared to correspond with median CMAP amplitudes, it was incongruent with MNCV findings. Consistent with our observations, Hattori et al. and Manganelli et al. reported that CMAP amplitudes, rather than MNCV values, are more closely associated with clinical severity in CMT.26,27 Ulnar sensory nerve action potentials (SNAPs) could also serve as an electrophysiological measure of disease severity. However, their frequent absence in the early stages significantly limits their utility in distinguishing severity levels between patients. 28 This limitation was evident in our study as well, preventing a meaningful comparison between the two demyelinating CMT groups.
All seven male patients with CMT1X included in this study exhibited an age at onset ranging from 6 to 45 years. Except for the patient carrying the novel GJB1:c.284T > C variant, the others, whose variants have been previously reported, exhibited typical CMT1X features: MNCVs nearly within the range of 25–45 m/s, reduced CMAP amplitudes in the upper extremities, and distal limb weakness with a maximum of MRC grade of 3/5. The 57-year-old patient with the GJB1:c.284T > C variant was the oldest in the CMT1X group and uniquely presented with severe plantar flexion weakness (MRC grade 0/5), and unexcitable nerves in NCS. His more severe phenotype may be attributed to his advanced age. Panosyan et al. demonstrated a strong correlation between age and disease burden in CMT1X, particularly in males. 29 Specifically, older males exhibit more pronounced reductions in motor and sensory neurophysiology parameters compared to younger males. Additionally, recent longitudinal analyses indicate that in CMTX1 patients, Charcot–Marie–Tooth disease examination score increases over time, with significant progression observed up to eight years of follow-up. 30
We identified two recessive mutations: PRX and IGHMBP2 (Table 2). Notably, the carrier of the PRX mutation also harbored an IGHMBP2 variant. Compound heterozygotes for the IGHMBP2 were detected in two cases that had tested negative in MLPA and the targeted sequencing of 11 genes in a previous study conducted in Vietnam (unpublished data). 19 One case had two pathogenic variants, NM_002180:c.1813C > T (p.Arg605Ter) and NM_002180:c.1334A > C (p.His445Pro), and the other had one pathogenic and likely pathogenic variant, NM_002180:c.1813C > T (p.Arg605Ter) and NM_002180:c.1015_1020del (p.Leu339_Glu340del), respectively. Notably, there was one Vietnamese family out of 13 families in the first observation of CMT2S-IGHMBP2 by Cottenie et al. (2014). 31 These data indicated that IGHMBP2 is the most prevalent recessive causative gene of CMT in Vietnam, accounting for 4.7% (7/150) of alleles across both studies. The two cases with IGHMBP2 compound heterozygotes died before the age of five due to long-term mechanical ventilation dependency, a clinical picture consistent with distal hereditary motor neuronopathy-1, which can overlap with the “classic CMT” phenotype.5,32 Contrarily, the IGHMBP2 carriers in our cohort exhibited characteristics consistent with CMT2S.5,31 The two IGHMBP2-related phenotypes were grouped by ClinGen owing to their shared mechanisms and inheritance patterns, both in vivo and in vitro. 33 Given the challenges in differentiating hereditary neuropathies with significant clinical and genetic overlaps, as in this case, Laurent Magy et al. proposed a new classification system to address the limitations of the phenotype-centric classifications that could hinder progress in research due to heterogeneous data.5,9,34
The patient diagnosed with CMT4F-PRX experienced lower-limb weakness from birth and began walking at three years of age. Over time, the motor weakness progressed to the upper limbs by age 31. At enrollment, muscle strength in the upper and lower limbs was rated 4/5 and 2/4, respectively. In addition to generalized areflexia, muscle wasting and sensory deficits were markedly more pronounced in the lower limbs compared to the upper limbs. NCS and EMG revealed chronic progressive demyelinating patterns consistent with the manifestations of homozygous PRX variants reported by Uchôa Cavalcanti et al.. 20 In our study, the autosomal recessive inheritance pattern accounted for 5.7% (2/35) of the genetically confirmed patients. When combined with data from the study conducted by Trung Hieu et al., this proportion increased to 8.3% (4/48). 2 This prevalence aligns with findings from populations with low consanguinity rates.3,5,7,19,20 Contrarily, regions in Africa wih high consanguinity rates exhibited a proportion of autosomal recessive CMT exceeding 90%. 2
In our study, the two panels of 94 genes yielded a 43.2% detection rate of pathogenic and likely pathogenic variants (19/44). This is a significant increase compared to the detection rates of 9.7% among the Vietnamese population reported by Trung Hieu et al. (2022), 30.7% among the Japanese population, and 24.4% among the Han Chinese population.3,4,19 This difference cannot be solely attributed to the size of the gene panel. If our study utilized a panel identical to that of Trung Hieu et al., which included only 11 genes (PMP22, MPZ, EGR2, NEFL, MFN2, GDAP1, GARS, MTMR2, GJB1, RAB7A, and LITAF), the diagnostic yield would have reduced to 34.1% (15/44). Despite this reduction, this yield remains nearly four times higher than that of the study by Trung Hieu et al., and half as much again as those of the studies by Y. Higuchi and H. Takashima despite their larger gene panel of 100 genes.4,19 Another factor that could dramatically increase or decrease the diagnostic yield of genetic testing is the criteria for patient selection, which can vary between studies. However, our patient selection criteria are aligned with those of Trung Hieu et al. (2022), while the criteria used in the studies by Y. Higuchi and H. Takashima were not well described. This lack of detailed inclusion criteria made it challenging to determine whether differences in patient selection contributed to the higher diagnostic yields reported in the studies.
A potential explanation for the discrepancy in diagnostic yield lies in the implementation of VarSome, which allowed the fulfillment of more pathogenic ACMG criteria, particularly those derived from larger populations and third-party pathogenicity predictors. Although bioinformatics tools such as VarSome have been used to enhance efficiency as part of a holistic classification approach, it is essential to recognize that pathogenic classification may vary across different tools due to input data variations. To address this issue, genetic reports should include fulfilled classification criteria to improve the consistency among laboratories and ensure that criteria are modified when necessary.35–37 Another cause of the discrepancy is that we conducted variant classification two years later, at which time more genetic information had become available, thereby increasing the cumulative diagnostic yield. 21 A similar phenomenon was observed in a study by Gregory et al., in which over 10% of explanatory variants were detected during reclassification after a two-year period. 38 We recommend the application of the bioinformatics platforms with periodic reanalysis to improve the yield of genetic testing, taking into account reporting criteria to avoid conflicts between laboratories.
Limitations
Our study has four main limitations. First, functional testing of variants is unavailable in Vietnam, and trio sequencing was not performed in all cases. Consequently, this resulted in unobtainable data, including cis/trans compound heterozygosity (PM3, PP1, BS4, BP2), definite de novo status (PS2), and functional impacts (PS3/BS3), which may result in inaccurate ACMG 2015 classification. Second, the budgetary constraints of our study precluded us from conducting further testing recommended by guidelines, including sequencing of untranslated regions beyond our mentioned techniques, variants of SORD and mitochondrial genes (e.g., MT-ATP6), CNVs, and whole-exome/whole-genome sequencing in patients with negative findings of MLPA and targeted NGS. 12 Third, our protocol did not include latency measurements of the blink reflex or facial nerve to distinguish the electrophysiological subtypes of CMT in the case of unmeasurable MNCV.12,21 Finally, assessing disease severity solely through the total MRC score fails to capture the comprehensive spectrum of clinical outcomes. Therefore, future studies should employ additional measurement tools such as the Overall Neuropathy Limitations Scale, CMT Examination Score, CMT Health Index, or CMT Functional Outcome.
In conclusion, a data-driven sequential genetic testing approach for CMT is more suitable in resource-limited regions. In studies conducted in Vietnam, MLPA provided no benefits for patients with axonal CMT. The combination of MLPA and targeted NGS achieved a diagnostic yield of 79.5%. NGS identified three novel pathogenic and likely pathogenic variants in GJB1, INF2, and IGHMBP2, as well as three novel VUS in MPZ, PMP22, and INF2. IGHMBP2 was the most prevalent autosomal recessive gene associated with CMT in Vietnam.
Footnotes
Acknowledgments
We extend our heartfelt gratitude to the neurophysiologists and technologists at the neurophysiologic laboratories of Military Hospital 175, University Medical Center of Ho Chi Minh City, and Children's Hospital 2 for their invaluable support in providing reliable NCS/EMG results. We also appreciate the collaboration with the team at the Medical Genetics Institute, whose meticulous efforts in double-checking uncertainties ensured the highest accuracy of the genetic results. Additionally, we acknowledge the Center for Molecular Biomedicine at the University of Medicine and Pharmacy at Ho Chi Minh City for their essential assistance in implementing MLPA in this study. Finally, we are deeply indebted to our patients, whose participation and motivation inspired this research, paving the way for advancements in the diagnosis and management of CMT.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability
The data supporting the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.