
Abstract
Background:
Phosphorodiamidate morpholino oligomer (PMO)-mediated exon skipping is currently used in clinical development to treat Duchenne muscular dystrophy (DMD), with four exon-skipping drugs achieving regulatory approval. Exon skipping elicits a truncated, but semi-functional dystrophin protein, similar to the truncated dystrophin expressed in patients with Becker Muscular dystrophy (BMD) where the disease phenotype is less severe than DMD. Despite promising results in both dystrophic animal models and DMD boys, restoration of dystrophin by exon skipping is highly variable, leading to contradictory functional outcomes in clinical trials.
Objective:
To develop optimal PMO dosing protocols that result in increased dystrophin and improved outcome measures in preclinical models of DMD.
Methods:
Tested effectiveness of multiple chronic, high dose PMO regimens using biochemical, histological, molecular, and imaging techniques in mdx mice.
Results:
A chronic, monthly regimen of high dose PMO increased dystrophin rescue in mdx mice and improved specific force in the extensor digitorum longus (EDL) muscle. However, monthly high dose PMO administration still results in variable dystrophin expression localized throughout various muscles.
Conclusions:
High dose monthly PMO administration restores dystrophin expression and increases muscle force; however, the variability of dystrophin expression at both the inter-and intramuscular level remains. Additional strategies to optimize PMO uptake including increased dosing frequencies or combination treatments with other yet-to-be-defined therapies may be necessary to achieve uniform dystrophin restoration and increases in muscle function.
ABBREVIATIONS
antisense oligonucleotides
Duchenne muscular dystrophy
extensor digitorum longus
immunoblotting
immunofluorescence
messenger RNA
phosphorodiamidate morpholino oligomer
relative standard deviation
tibialis anterior
quantitative real time PCR.
INTRODUCTION
Duchenne muscular dystrophy (DMD) is a progressive, X-linked neuromuscular disease caused by mutations in the DMD gene, resulting in out-of-frame mRNA transcripts and absence of functional dystrophin protein at the myofiber plasma membrane [1]. Exon skipping has been established as a promising therapeutic strategy for DMD, utilizing antisense oligonucleotides (AOs), such as phosphorodiamidate morpholino oligomer (PMO), to bypass mutated exons, and restore the mRNA open reading frame, yielding translation of a truncated, partially functional dystrophin protein. This corrective therapeutic strategy has proved successful in dystrophic animal models of DMD [2–5], confirming the rationale for human clinical trials and conditional approvals by the Food and Drug Administration (FDA) [6–9]. Approval of three of these drugs, Exondys 51 (eteplirsen), Vyondys 53 (golodirsen), and Amondys 45 (casimersen), were given to Sarepta Therapeutics, and NS Pharma, Inc. received approval for Viltepso (vitolarsen). Vyondys 53 and vitolarsen are both for patients who have a confirmed mutation of the dystrophin gene that is amenable to exon 53 skipping. Vyondys 53 was shown to increase dystrophin levels from 0.1%of normal at baseline to 1.02%of normal after 48 weeks of drug treatment. Viltepso was shown to increase dystrophin levels from 0.6%of normal at baseline to 5.9%of normal after 25 weeks of treatment [8, 9]. Exondys 51 is indicated for DMD patients with genetic mutations that are amenable to exon 51 skipping; however, clinical evaluation of eteplirsen, while promising in terms of its safety profile, still failed to meet primary endpoint measures [10]. As discussed above, FDA approval of these drugs is based on their ability to elicit exon skipping and dystrophin expression. Whether these drugs can improve clinical outcomes is not as clear [11]. As a result, the FDA recently released comprehensive guidance regarding eteplirsen citing considerable doubt regarding drug efficacy, clinical benefit, and the use of dystrophin as a primary endpoint measure. Most recently, Amondys 45 received approval for DMD patients who have a genetic mutation amenable to exon 45 skipping based on its ability to significantly increase dystrophin protein levels from baseline after 48 weeks of treatment [6]. These studies were difficult to interpret because of insufficient and highly variable AO-induced dystrophin expression, which, when compounded by the sampling limitations of minute muscle biopsies, has resulted in poor clinical outcomes and doubt regarding potential drug efficacy [2, 12–16]. Ultimately, these findings have led the FDA to question the relationship between dystrophin rescue and the functional benefit [10], stating there is a “disconnect between increased dystrophin expression and clinical efficacy.” Moreover, the FDA questioned current biochemical methods to quantify dystrophin levels accurately and reliably in response to treatment. The reasons behind the apparent disconnect between dystrophin expression and clinical efficacy remain unknown at this time and highlight the need to further investigate the mechanisms that regulate exon skipping therapies.
Previously, we reported high variability and sporadic localization of dystrophin restoration in response to a single high dose of PMO (1 month post-PMO injection) in the mdx mouse model of DMD [17]. This study showed that a high dose (800 mg/kg) of morpholino resulted in variable dystrophin rescue with no significant increase in force generation. In this study, we investigated the effects of chronic, maximal PMO dosing on muscle function and dystrophin restoration in mdx mice. We hypothesized that chronic treatment of mdx mice with high doses of PMO would normalize dystrophin expression and reduce inter/intra-muscular variability to achieve consistent improvement of muscle function. In DMD boys, exon skipping treatment must be chronically delivered, making it critical to understand both the short- and long-term effects of this treatment [16, 18]. To address this, we administered a series of monthly high doses of PMO (800 mg/kg) intravenously in the mdx mouse model and assessed dystrophin expression and muscle function after 6 months. Our data suggest that monthly, high dose PMO treatment does not reduce the variability that we have previously reported [17], however higher dystrophin levels were detected. Additionally, we see increases in specific force that were not improved in our short-term study. This taken together with the increased dystrophin protein, indicate dystrophin can potentially serve as a surrogate biomarker to predict a functional outcome in clinical trials.
MATERIALS AND METHODS
Animal treatment
All animal procedures were conducted in accordance with guidelines for the care and use of laboratory animals as approved by the Institutional Animal Care and Use Committee (IACUC) of Children’s National Health System. One month old male C57BL/10 ScSn:D mdx/J (mdx) mice were purchased from The Jackson Laboratory. Eight, 6-week-old mice received their first intravenous dose of 800 mg/kg PMO (5’-GGCCAAACCTCGGCTTACCTGAAAT-3’, GeneTools, LLC) in 300μL of total saline via the retro-orbital sinus. Body mass was measured the day before injection to determine the amount of PMO required in reference to the body mass for each mouse. PMO was prepared fresh on the day of injection. Intravenous administration of PMO was performed once for the acute treatment and endpoints were one month after the last injection [17]. For the long-term treatment, PMO was administered every 4 weeks for a total of 6 injections using insulin syringes with a 28 1/2 gauge permanently attached needle. Eight control mice were injected intravenously via the retro-orbital sinus with 300μL of saline. Male C57BL/10 mice (WT) were purchased separately and aged matched as controls for molecular and functional assays.
Voluntary Wheel running
After 4.5 months of treatment with PMO, mice were placed in isolated cages to measure voluntary wheel activity [19, 20]. Activity was measured for a total of 7 consecutive days of which the first 48 hours were considered a normalization period and these values were not included with the final data [21].
Grip strength
Forelimb and hindlimb grip strength parameters were performed blindly by a technician as previously described and in accordance with TREAT-NMD SOPs [22–25].
Open field Digiscan
Mice were acclimated in digiscan boxes for one hour each day for a week and testing was performed at the same time of day on the following week as previously described [22, 26].
In vitro muscle force
In vitro force measurements of the EDL muscle were performed at the time of euthanasia as previously described. In brief, mice were anesthetized and the EDL was surgically removed, and maximal force was measured. These measurements were determined for all mice, saline and PMO-treated. Cross sectional area of the EDL muscle was calculated to determine specific force. This was performed as previously described [24] and in accordance with the following TREAT-NMD SOP [27].
In vivo imaging of muscle inflammation
Mice were injected with ProSense 680 (2 nmol per mouse in 150μL 1X PBS, IP, PerkinElmer) 24 hours prior to imaging as previously described [23, 28–31]. The following day, mice were anesthetized, and fluorescence was determined for both hind and fore limbs. One mouse was used for each group to determine background and the day following injection, in vivo imaging was performed to determine the level of cathepsin cleavage in the mice.
Exon skipped transcript detection by quantitative real time PCR (qRT-PCR)
Total RNA was extracted from muscle using a standard TRIzol (Life Technologies) isolation technique. Purified RNA was reverse-transcribed using Random Hexamers and High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher). Relative skipped Dmd mRNA transcript levels were measured by qRT-PCR using specified TaqMan probes with Taqman Fast Advanced MasterMix on a 7900HT Fast Real-Time PCR system (Thermo Fisher). TaqMan probe for the skipped Dmd product (AIOIXIL; Thermo Fisher) was designed to amplify the splice junction at Dmd exons 22–24. For the non-skipped Dmd mRNA, TaqMan probe (Mm0126935_m1; Thermo Fisher) was used to amplify the region spanning Dmd exons 23–34. Percent exon skipping was calculated based on the corresponding DCt values for skipped and total Dmd transcript normalized to titan (Ttn) (Mm00621005_m; Thermo Fisher) and 18S (Mm03928990_g1; Thermo Fisher) mRNA, using the following [(Average of triplicate reactions of skipped Dmd)/(Average of triplicate reactions of skipped Dmd + Average of triplicate reactions of non-skipped Dmd)] * 100.
Dystrophin detection by Western blotting
Muscle lysates were prepared using RIPA buffer (Teknova) supplemented with 1X protease inhibitors. Samples were homogenized in chilled supplemented RIPA buffer on ice using a handheld motorized homogenizer. Protein assays were performed using the DC assay as described by the manufacturer (Pierce). Pieces of various muscles (quadriceps, triceps, gastrocnemius, TA, diaphragm, soleus, EDL, and heart) were homogenized by grinding the tissues with a mortar and pestle cooled on dry ice and placing the pulverized muscle in a high SDS lysis buffer supplemented with 1X protease inhibitors and β-mercaptoethanol. Protein assays were performed using either the Bradford assay for lysates in RIPA or BCA assay for those with high SDS lysis after treatment with a Compat-Able protein assay Preparation Reagent (Thermo) per manufacturer’s instructions [17].
SDS-PAGE Electrophoresis
Muscle lysates of equal total protein concentrations were prepared with reducing agent and loading buffer and loaded onto 4–8%Tris-acetate gels with molecular weight markers. Proteins were separated by gel electrophoresis at 150V for one hour and subsequently transferred to nitrocellulose membranes at 4°C for 75 minutes at 0.3A. After transfer, membranes were stained with Ponceau solution to determine efficiency of transfer. Membranes were washed in TBST and then blocked with 5%(W/V) non-fat milk in TBST for one hour. Primary antibodies were prepared in 5%milk TBST and added to membranes overnight at 4°C. Dystrophin was detected using the mouse anti dys1 and dys2 antibodies (Lecia, 1:1000 dilutions). Mouse anti-Vinculin was used as a loading control (1:3000, Abcam). The following day, membranes were washed three times in TBST prior to application of the secondary antibodies (Rabbit anti Mouse: 1:3000, 1:5000 dilutions in 5%milk in TBST) for one hour at room temperature. Blots were developed using ECL and film for visualization. Quantification of densitometry was performed using ImageJ.
Detection of PMOs in muscle by ELISA
PMO levels in muscle lysates were analyzed as previously described [32]. In brief, sample lysates were diluted 1/20, 1/200, and 1/2000 in a control muscle lysate buffer (0.2 ug/μL protein), and PMO standards were diluted to various concentrations in a similar manner. Hybridization was facilitated using an anti-sense probe to the PMO with both a biotin epitope and a DIG tag for hybridization and carried out at 37°C. After hybridization, 100μL of the hybridization mix was pipetted into duplicate wells on avidin-coated plates and incubated at 37°C for 30 min. The plates were then washed, and each well was treated with micrococcal nuclease (NEB, M02475), followed by incubation with an anti-digoxigenin-AP Fab fragment antibody (Roche, 11093274910). Lastly, AttoPhos Substrate (Promega, S101C) was added to the plates and incubated at 37°C for 30 min. Fluorescent readings were obtained, and PMO concentrations were quantified and calculated on the basis of the PMO standard curve.
Immunofluorescence
Frozen muscle tissues were sectioned at 10-μm thick and stored at –20 °C until used. Immunofluorescent (IF) for dystrophin protein was performed as described previously [17]. In brief, the muscle sections were brought to room temperature (RT) but not fixed. For dystrophin staining, unfixed sections were blocked with 10%normal sheep serum, followed by incubation overnight at 4°C in a humidified chamber with a P7 dystrophin antibody (1:400; Fairway Biotech, England). Next, the sections were washed and probed with goat anti-rabbit IgG Alexa 594 antibody (1:300; Life Technologies, Grand Island, NY, USA) at RT for 1 h and counterstained with 4′,6-diamidino-2-phenylindole (DAPI) for nuclear staining. The stained tissue sections were stored at 4°C for further imaging and quantification analyses. Staining was confirmed using alternative dystrophin antibody (Genetex, Irvine, CA, USA). Images were acquired using an Olympus BX61 microscope with attached Olympus DP71 camera module. The surface area of each section and the relative proportion of the dystrophin-positive fiber area were determined using ImageJ software. Analysis and quantification were performed using cellSens 1.13 (Olympus), MetaMorph Premier 7.7.0.0 (Molecular Devices, San Jose, CA, USA), Adobe Photoshop CS6 (Adobe, San Jose, CA, USA), and ImageJ (NIH, Bethesda, MD, USA) software.
RESULTS
Dystrophin is restored after chronic PMO treatment
Five-week-old mdx male mice (n = 8) were systemically treated with a high dose of PMO (800 mg/kg) injected via the retro-orbital sinus once per month for six months (chronic treatment). Percent exon skipping and dystrophin protein expression relative to WT were quantified in several muscles, including quadriceps, triceps, gastrocnemius, TA, diaphragm, soleus, EDL, and heart. Dystrophin transcripts were compared to the unspliced transcripts using probes targeting the exon 23-24 junction, which was in all muscle samples. Dystrophin was detected from PMO treated mice (EDL, quadriceps, gastrocnemius, diaphragm, and triceps), while skipped transcripts were not detected in any saline treated controls (Supplemental Figure 1).
Dystrophin protein restoration was observed by immunoblotting (IB) in all skeletal muscles and diaphragm in mice administered PMO, with the heart expressing the lowest amounts of dystrophin at an average of 3%(range 0.44%to 4.63%, n = 8) (Fig. 1a-b). The tibialis anterior (TA) had the highest levels of dystrophin protein expression at an average of 40%(range 23.7%to 60%, n = 8). The soleus had the lowest levels dystrophin protein expression at an average of 1.15%(range 0.03%to 5.4%, n = 8). Quadriceps, gastrocnemius, and triceps averaged at 7%, 12%and 18%of WT dystrophin levels, respectively. The soleus had low and highly variable dystrophin protein levels with the relative standard deviation (RSD) at 155%. The quadriceps showed the next highest level of variability among the eight mice tested, with an average (mean± SD) of 6.9%±7.83%(RSD of 113%). The TA had the lowest variability with an average dystrophin level of 40.4%±12.9%(RSD of 32%) (Table 1). For all muscles except the triceps, as the average dystrophin amount increased, the RSD decreased, highlighting the challenges associated with quantifying low abundance proteins.
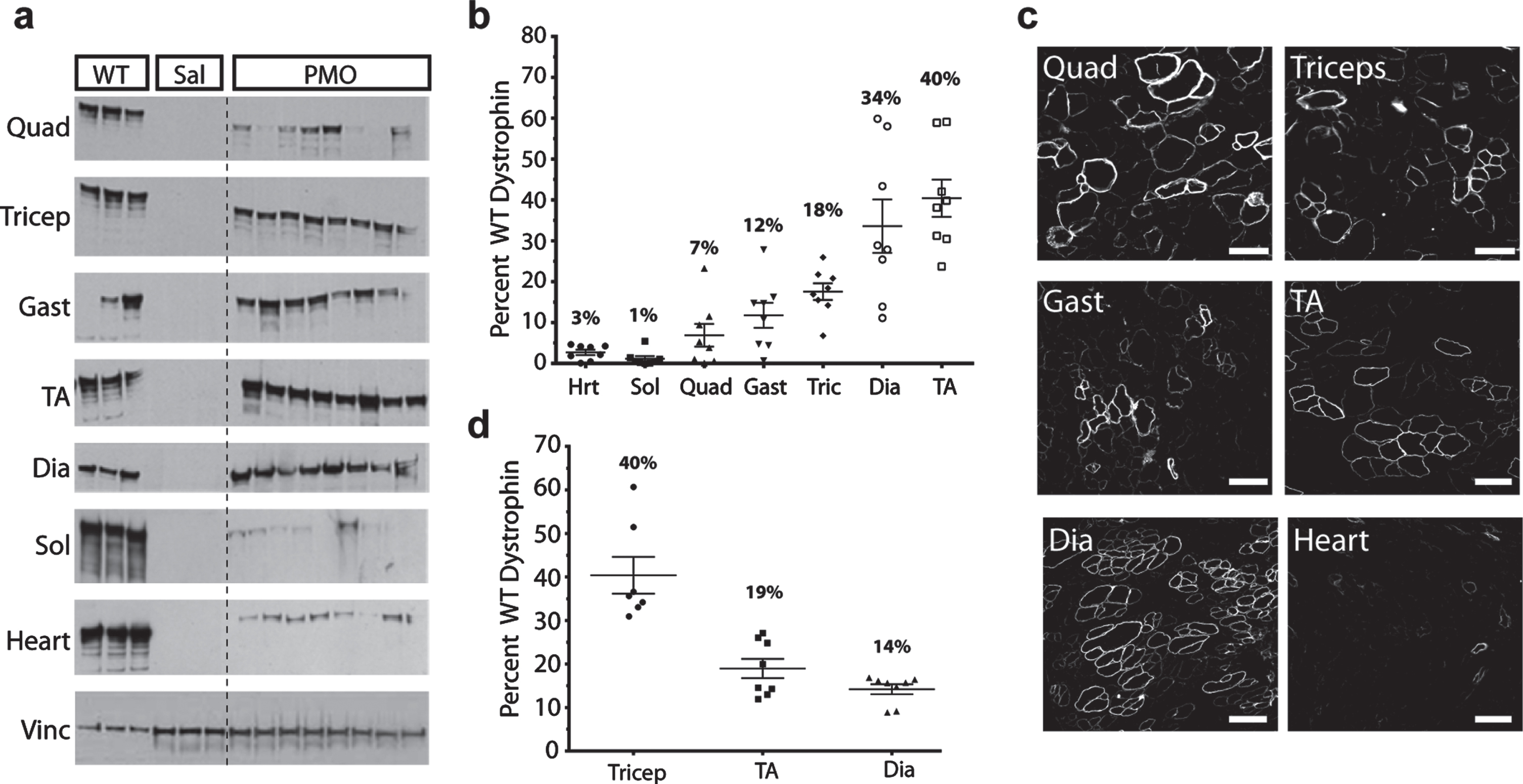
Chronic PMO treatment increases dystrophin protein expression in skeletal muscles. (a) IB of dystrophin protein from quadriceps (quad), triceps (tric), gastrocnemius (gast), tibialis anterior (TA), diaphragm (dia), soleus (sol), and heart (hrt) in PMO-treated mdx mice (n = 8) vs. saline and WT control. Note that dystrophin protein is restored in skeletal muscles and to a lesser extent in the heart after chronic PMO treatment. Blots show variability of dystrophin expression between muscles in PMO-injected mdx mice. Dystrophin was detected as a band at approximately 427 kDa. Vinculin at 117 kDa was used as a loading control. WT samples were loaded with 25μg total protein while saline and PMO-injected samples were loaded at 75μg of total protein. For the gastrocnemius, the most abundant WT sample was used for analysis. For the TA sample the two most abundant WT samples were used for the analysis. For all other muscles, the average for the three WT samples was used for the analysis. Densitometric analysis was performed using Quantity One software. (b) Dystrophin quantification by IB, demonstrating the percentage dystrophin expression in PMO injected mice vs. WT (set to 100%). Plots show variability between mice within a muscle group and between muscles. (c) Representative images of an individual mouse showing variability between all muscles. Images were selected to show positive fiber clustering and do not represent total area imaged and quantified. All tissues were sectioned stained, and probed with goat anti-rabbit IgG Alexa 594 antibody. Dystrophin positive fibers were normalized to the area of the muscle section and the WT percentage of positive fibers. Original magnification for c = x40; scale bar, 100μm. (d) IF quantification of triceps, TA, and diaphragm for all mice (n = 8).
Dystrophin quantification in skeletal muscles by IB (%relative to WT)
Dystrophin positive fibers were quantified by IF in three muscles (triceps, TA, and diaphragm). When examining all muscles from an individual mouse, we found dystrophin positive fibers to be grouped into clusters, geographically dispersed across the entire muscle section, as previously reported (Fig. 1c and Supplemental Figure 2) [17]. The percent positive dystrophin fibers in the triceps, TA, and diaphragm was 40%(range: 31–61%), 19%(range: 12–26%), and 14%(range: 9–17%) respectively (Fig. 1d). By IF, dystrophin quantification was found to be less variable, as evidenced by the decreased RSD values (∼30%for all muscles analyzed) (Table 2). Overall, dystrophin protein expression levels after chronic PMO treatment were higher than that of the acute treatment [17]. However, dystrophin protein quantification revealed that protein rescue levels remained variable, with quantification methods (IF and IB) showing poor agreement.
Dystrophin quantification in skeletal muscles by IF (%relative to WT)
Sustained dystrophin expression by exon-skipping increases body weight
At the start of treatment, body weight was similar for saline-treated controls and PMO-treated mice. The same was observed at the beginning of our acute, one month treatment [17]; however, no difference in body weight was detected at the end of the acute treatment (data not shown). Similarly, one-month post chronic PMO administration, no changes in body weight were detected (Supplemental Figure 3). By three months of age the PMO-treated mdx mice had slightly increased body weight (5%), a trend that continued throughout the remainder of the study (Supplemental Figure 3). At the end of the study, PMO-treated mdx mice continued to trend towards increased body weight compared to saline-treated controls.
Behavioral analysis began with voluntary wheel testing at 4.5 months for one week, while grip strength acclimation began at 5.5 months. EDL maximal force measurements were performed on the day of euthanasia. Interestingly, we observed a decrease in body weight for both saline control and PMO-treated mdx mice during this testing period. The decline in weight was less in PMO-treated mice than in saline control (PMO treated 2%vs. control 4%decrease) (Supplemental Figure 3).
After euthanasia, individual muscles were wei-ghed. Consistent with the increase in body mass observed in vivo, PMO-treated muscle weights increased significantly compared to saline controls. The muscles examined and percent weight increase over saline included: quadriceps (14%), triceps (13%), gastrocnemius (10%), and TA (10%) (Supplemental Figure 4a-d). The increase observed in the TA was not statistically significant. The diaphragm and soleus showed no muscle weight difference between the groups (Supplemental Figure 4e, f). In addition, various organs including the heart, lungs, kidneys, liver, and brain (Supplemental Figure 5) showed no changes in weight. There was no difference in muscle weight for the EDL (which is the muscle tested in the force generation protocol) between PMO-treated and saline controls at the end of the acute or chronic treatment (Fig. 2a). There was a significant increase in the size of the muscles when comparing the mdx mice (administered saline or PMO) compared to WT controls (Fig. 2a). Sustained dystrophin expression by exon skipping resulted in increased body weight and increased muscle mass of some skeletal muscles.
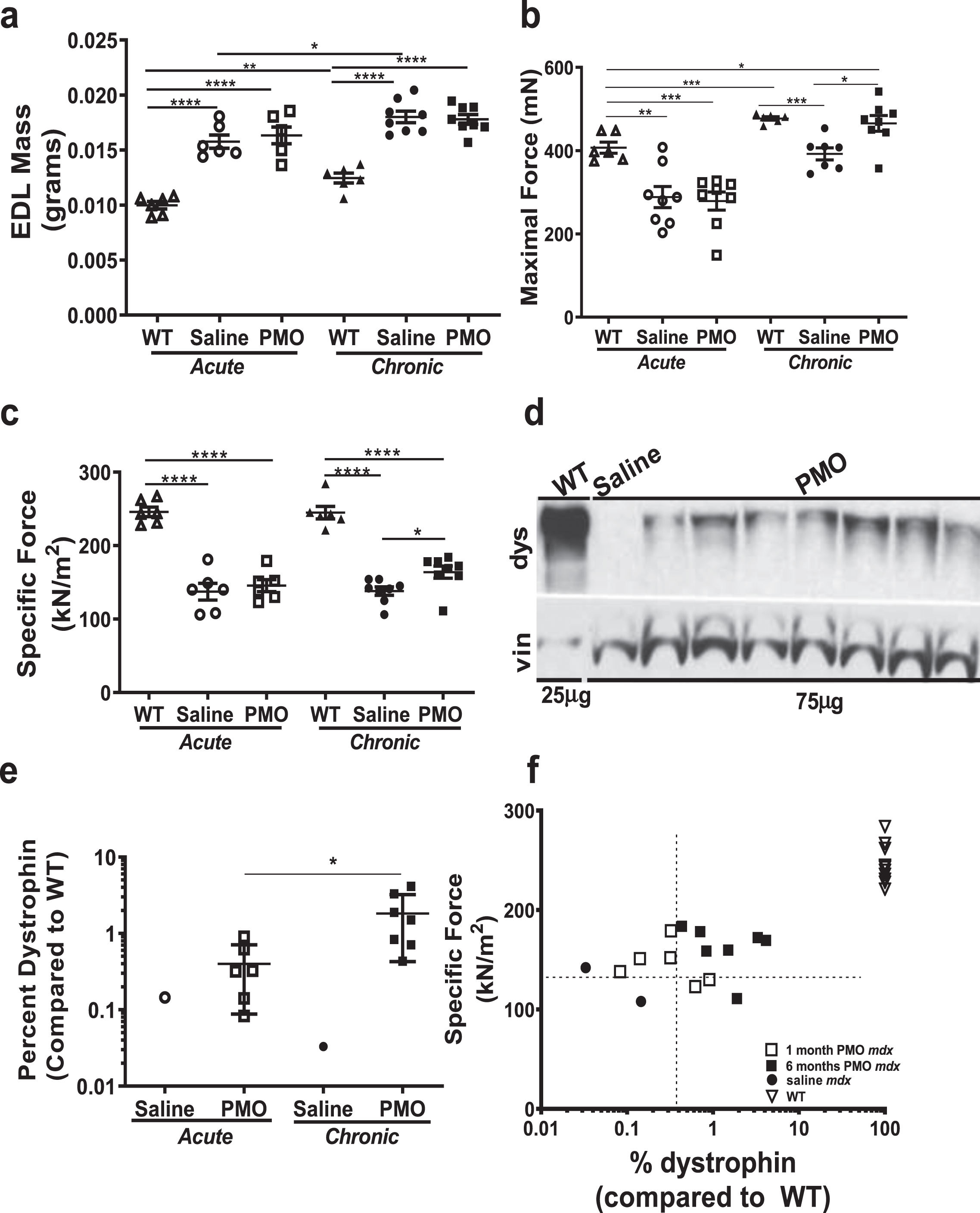
Muscle force improves after chronic PMO administration in mdx mice. Muscle force generation by EDL muscle was measured after both acute and chronic PMO treatment and compared to age-matched saline controls and WT. (a) EDL muscle weights (mg) at the end of acute and chronic treatments. (b) Maximal force (mN) after acute and chronic treatments. Note that after acute treatment force is not changed between PMO and saline treated mdx mice, while WT force is significantly increased. However, after chronic treatment, maximal force is significantly increased (35%; p < 0.05) in PMO mice as compared to saline controls. (c) Specific force (kN/m2) also shows significant increase only after chronic PMO treatment. (d) Immunoblotting and (e) densitometry quantification for dystrophin protein in the EDL muscle lysates after chronic PMO treatment. (f) Correlation plot between dystrophin expression and force. Student’s t test (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001). (dys –Dystrophin, 427kDa, vin –Vinculin, 117kDa, WT-C57BL10).
Dystrophin expression threshold for muscle force improvement
In vitro force measurements were performed on the EDL muscle at the end of the study to determine the force generation capacity of skeletal muscle after chronic PMO administration. There were no differences in maximal or specific force in mdx mice administered PMO compared to saline controls after the acute treatment (7%and 5.6%increase in the PMO group respectively) (Supplemental Figure 6). However, after chronic treatment there was a significant increase in maximal force (35%) in the mice administered PMO chronically compared to saline controls (Fig. 2b). Specific force (maximal force normalized to cross-sectional area of the EDL muscle, kN/m2) was significantly increased (19%) in mice administered PMO mice compared to saline (Fig. 2c); however, specific force still did not reach WT levels after either acute or chronic treatment.
Dystrophin protein expression was restored in the EDL muscles in the mice administered PMO chronically (Fig. 2d). After acute treatment, dystrophin rescue in EDL muscles was on average 0.3%of WT. After chronic treatment, this percentage increased to 1.83%(Fig. 2d, e), which was enough to show improvement in muscle function (as shown in Fig. 2c). Percent dystrophin was plotted against specific force measurements from individual mice (Fig. 2f). The PMO-treated mdx mice had the greatest specific force and all but one animal had a force greater than 150 kN/m2. Mice from the acute study also grouped together, showed a threshold force of less than or equal to 150 kN/m2. Saline treated mice from both studies had a specific force measurement lower than 150 kN/m2. These results indicate there may need to be a certain threshold of dystrophin expression restored before improvements in force can be significantly achieved. However, further experiments would have to be performed to determine the exact threshold amounts of dystrophin required for functional improvements in a clinical setting. Here we show that one high dose of 800 mg/kg is not sufficient to improve muscle strength, but repeated PMO injections result in increased muscle force, highlighting that exon skipping improves function in a dose-dependent manner.
Behavioral outcomes are not changed after chronic PMO treatment
Grip strength measurements were not significantly increased in mdx mice administered PMO compared to saline treated controls. Absolute forelimb measurements showed a 10%increase (Fig. 3a), which when normalized showed a 6%increase (Fig. 3b). Absolute hindlimb increased by 7%(Fig. 3c), while normalized hindlimb fell to only a 2%increase (Fig. 3d). Although grip strength trended towards an increase in the PMO treated group, this was not statistically significant (Fig. 3).
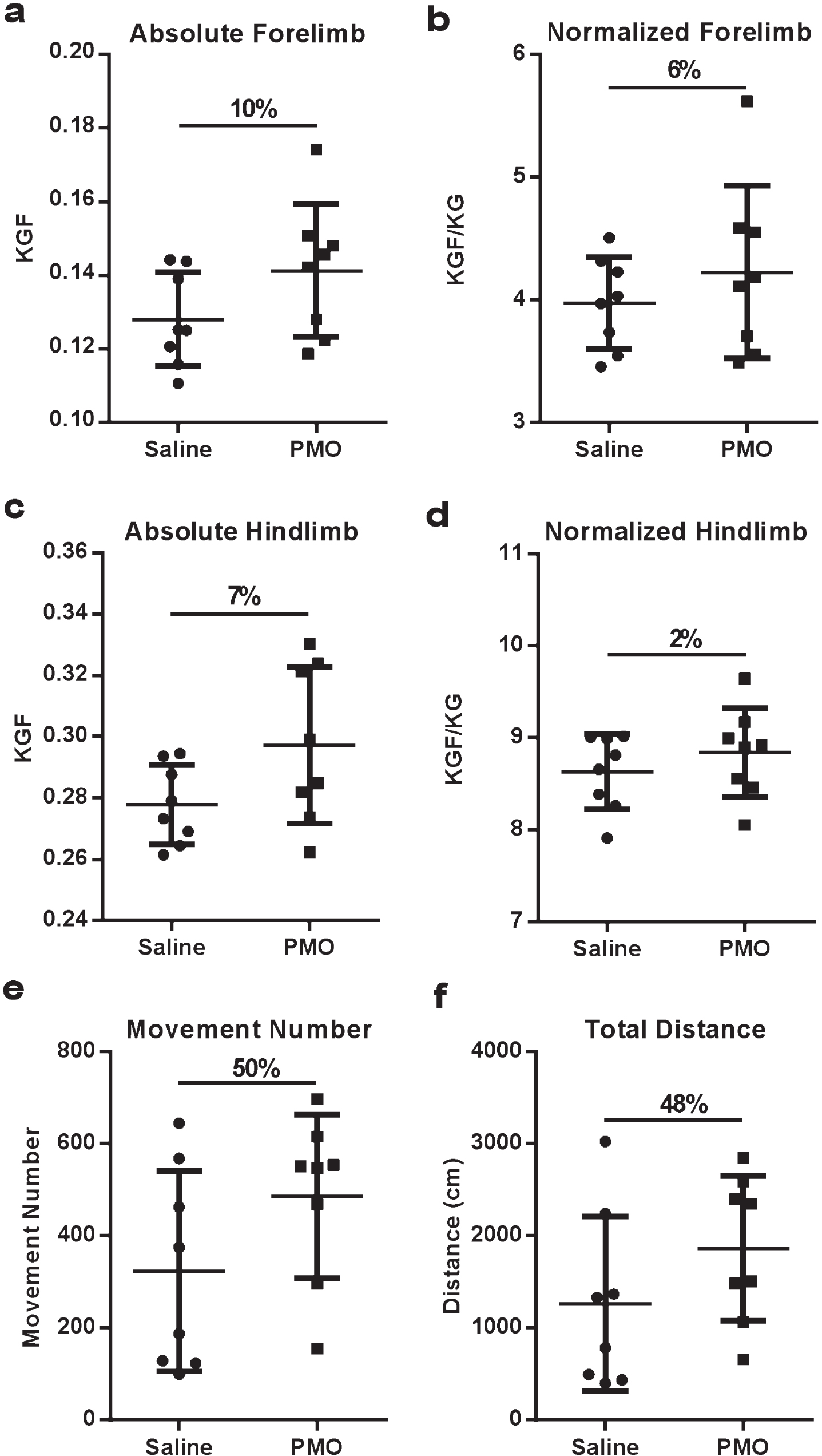
Behavioral outcomes are not significantly increased after chronic PMO treatment. Although overall activity was increased in the PMO-treated cohort, this was not statistically significant. (a) Absolute forelimb measurements trended toward increasing in PMO treated mice. (b) Forelimb strength normalized to body weight is also increased in PMO treated mice. (c) Absolute hindlimb measurements were increased by 7%in PMO treated mice. (d) Normalized hindlimb measurements were not changed in PMO treated mice (2%increase). (e) Movement number was increased 50%in mdx mice treated with PMO. (f) Total distance was increased 48%in PMO treated mdx mice. Data presented as mean± SEM. Statistical analysis was performed using Student’s t test.
Digiscan analysis showed that PMO-treated mice had a 50%increase in number of movements (Fig. 3e), 48%increase in total distance (cm) (Fig. 3f), and a 53%increase in movement time (measured in seconds) (Supplemental Table 1). Also, horizontal activity was approximately 34%greater in PMO-treated mice (Supplemental Table 1). Accordingly, the PMO-treated mice showed a decrease in the rest time (Supplemental Table 1). Finally, voluntary wheel analysis showed no change in any of the measurements which included: total distance run (km), average speed (km/hr), average speed when running (km/hr), and max speed (km/hr) (Supplemental Table 2). None of the behavioral outcomes were statistically significant, although they all trended towards increased behavioral activity in mice administered PMO.
Dystrophin rescue does not correlate with residual PMO concentration in muscle tissue
Residual PMO in skeletal muscle lysates from chronic PMO treatment were measured by hybridization ELISA. After 6 months, there were very low levels of PMO in the triceps and gastrocnemius muscles (n = 3 each group, Fig. 4a). When compared to PMO concentration levels after acute treatment, chronic levels were found to be even lower, although less variability was observed (Fig. 4b). There was no correlation between PMO levels and dystrophin rescue, as reported previously (Fig. 4c) [17]. This indicates that chronic, monthly dosing does not increase accumulation of PMO in the muscle even at the high dose levels used in this study.
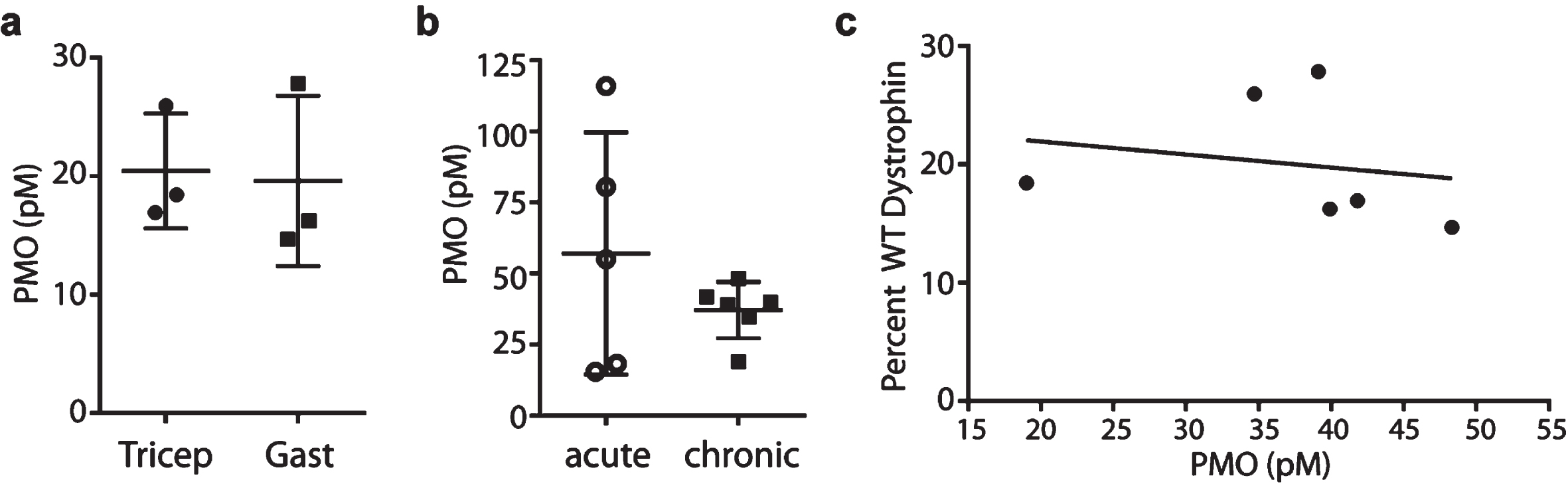
Residual PMO in skeletal muscles after repeated PMO injections. (a) Residual PMO levels measured by hybridization ELISA in triceps and gastrocnemius muscles at the end of chronic PMO treatment. (b) Comparison between PMO levels after acute and chronic PMO treatments. Note that the average PMO concentration in muscle is lower after chronic treatment, yet variability is decreased. (c) Percentage of WT dystrophin as measured by IB and PMO levels measured by hybridization ELISA were plotted with a regression line and showed no significant correlation.
Dystrophin expression variability is improved with increased frequency of PMO injections
To investigate whether increased frequency of PMO injections would normalize the variable rescue of dystrophin, we treated 4-week-old mdx mice with 25 or 100 mg/kg PMO IV once a week for 4 weeks. Dystrophin protein expression analyzed by IB was still variable with the 25 mg/kg regimen in the triceps. Two of the four mice showed high amounts of dystrophin protein, while no bands were detected for the other two animals. Interestingly, when mice were administered PMO at 100 mg/kg once per week, dystrophin protein levels were similar across all treated animals, as evidenced by the similar band sizes (Supplemental Figure 7). Specific force increased by 16%in the mice administered the weekly, higher PMO dose (100 mg/kg), when compared to saline treated mice and the mice administered 25 mg/kg PMO weekly had a 9%increase in specific force (Supplemental Figure 7).
Inflammation in forelimb muscle is decreased in PMO treated mice
Muscle inflammation in vivo was quantified using live animal optical imaging to measure photon intensity (an indicator of cathepsin enzyme activity) in the limb muscles of mdx mice, using ProSense-680 dye. PMO treatment significantly reduced muscle inflammation in the forelimb (p < 0.01); however, this effect was not observed in the hindlimb (Supplemental Figure 8a-d) muscles. Decreased inflammation in triceps muscle was confirmed using H&E staining (Supplemental Figure 8e-f).
DISCUSSION
Repeated PMO administration increases the success of exon skipping; however, high dose levels are required to obtain widespread and therapeutically relevant levels of restored dystrophin [2, 4]. The variable dystrophin rescue levels observed in both pre-clinical and clinical trials highlight the need for further optimization of exon skipping therapies. We tested whether repeated high dose injections of PMO would normalize the dystrophin rescue variability observed in exon skipping trials. After chronic, high dose PMO treatment, we quantified dystrophin protein in various muscles and measured force generation. Six monthly administrations of PMO at 800 mg/kg induced expression of dystrophin protein and resulted in functional improvements. The amount of protein quantified here was higher than that of the acute treatment; however, the pattern of dystrophin expression was still variable, with dystrophin positive fibers observed in clusters dispersed throughout the muscle sample. It should be emphasized that the PMO used here targets exon 23 of the mouse dystrophin gene which is different than the PMOs targeting human dystrophin exons; therefore, making direct comparisons between preclinical models in mice and clinical trials in humans difficult. Given the challenges associated with interpreting clinical data due to small sample size and lack of a clear relationship with positive functional outcomes, these models provide preliminary evidence for dystrophin rescue using exon skipping in the mdx mouse, and supports human clinical findings using PMOs.
Dystrophin expression was detected in all skeletal muscles of PMO-treated mdx mice, with individual muscles reaching up to 60%of WT levels in the TA, with an average of 40%. Notably, the average of dystrophin rescue for other skeletal muscles (quadriceps, triceps, gastrocnemius, soleus, EDL) was below 20%of WT. After acute PMO treatment, we reported that the highest levels of dystrophin rescue by WB were observed in the triceps muscle; however, after chronic treatment the TA muscles were the highest responders. Additionally, the TA muscles showed the highest level of variability, and the lowest variability was observed in the diaphragm muscle after acute treatment. After chronic PMO administration, however, the quadriceps showed the highest variability (after soleus) among all PMO-treated mice, with dystrophin protein varying from 0%to 23%of WT levels. There was no correlation between residual PMO in muscle and dystrophin expression after chronic PMO administration, as seen with our previous results after 2-, 7-, and 30-days of treatment, [17]. PMO levels were lower after chronic treatment than 30 days post-acute treatment, albeit less variable. The present findings reinforce the conclusion that the effects of exon skipping are variable and the biochemical factors driving dystrophin expression variability need to be elucidated to improve therapy.
Systemic delivery of AOs to dystrophin-deficient muscle is known to be dose-dependent [2, 33–35]. Previous studies have reported an increased, but still variable, dystrophin expression following weekly PMO administrations for 4 weeks [34]. For this study we tested high doses administered monthly because exon skipping will likely be a chronic treatment for DMD patients and minimizing the number of injections required for the treatment regimen would be preferable.
Regenerating muscle fibers are variable throughout mdx disease progression and could impact dystrophin protein rescue [35]. We previously showed that robust and efficient PMO uptake into muscle occurs in areas of myofiber repair where PMO localization is sustained in inflammatory loci, enters macrophages, and is subsequently delivered to myoblasts and newly formed myotubes [35]. Our data demonstrates that inflammation is decreased in forelimb muscle but not in hindlimb muscles despite gene correction. The impact of ongoing muscle inflammation on new dystrophin expression at mRNA levels is unknown.
Variable dystrophin rescue could also be due to inflammatory or dystrophin-targeting microRNAs (miRNAs). Our group has previously demonstrated that pro-inflammatory cytokines such as tumor necrosis factor alpha (TNFα) control dystrophin-targeting miRNAs. These miRNAs regulate dystrophin expression, demonstrating inflammatory miRNAs can contribute to variable dystrophin levels in muscular dystrophy [36]. One mechanism to control pro-inflammatory miRNAs is administration of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB) inhibitor drugs such as Vamorolone. This type of drug in combination with PMOs has the potential to enhance dystrophin restoration in disease models [24, 37].
The current standard of care for DMD patients includes the use of corticosteroids; however, this does not address the genetic component of the disease. Therefore, the potential impact of combination therapy using exon skipping and corticosteroids, is an important consideration given that DMD patients may benefit the most from a multidrug approach. Although the current study did not examine this combination treatment approach, it has previously been shown that prednisolone does not affect exon-skipping in patient-derived muscle cells treated with AOs and may enhance dystrophin expression in mdx mice [38].
Chronic exon skipping therapy resulted in skeletal muscle function improvement, as measured by in vitro force generation by EDL muscles. We observed that the weight of EDL muscles were greater in the mdx mice (PMO and saline treated) compared to WT at the end of both acute and chronic treatment. After chronic treatment, PMO-treated mdx mice (but not saline controls) had maximal force comparable to WT. However, when analyzing specific force, the cross-sectional area of the muscle is factored in and because mdx mice had larger muscles, specific force failed to reach WT levels. Nevertheless, specific force of PMO-treated mice was significantly increased compared to saline-treated controls (18.7%increase). After acute treatment, specific force generation was not significantly increased (5.9%increase compared to saline control). To test whether this increase in force after chronic treatment correlates with amount of dystrophin rescued in muscle, we quantified amount of protein in EDL muscles by IB. After chronic PMO treatment, dystrophin expression in EDL muscles was on average 1.8%of WT, while after acute treatment the average was only 0.3%. Although dystrophin protein levels were very low, this 6-fold increase in dystrophin was sufficient to cause muscle function improvement. This is in accord with an earlier report showing that changes in muscle strength are related to dystrophin expression levels, as induced by repeated doses of peptide conjugated PMO [39]. In this study, average dystrophin expression of 15%of WT muscle levels was necessary for in situ functional improvement in the TA muscle. Linear regression analysis showed a positive correlation between maximal specific force and dystrophin protein expression. Interestingly, individual muscles showed no correlation between force and dystrophin expression, indicating the highest dystrophin expressing muscles did not generate the highest force. This lack of correlation may result from the low sensitivity of IB for quantifying low-abundance proteins. In addition, the small size of the EDL muscles poses a challenge when extracting protein for analysis. This highlights the need for having robust and reliable measures of dystrophin quantification utilizing standardized procedures and multiple testing methods [40]. These concerns are also problematic in clinical trials of exon skipping, where low dystrophin levels have been achieved with the current AOs from small muscles biopsies that might not be representative of the entire muscle.
Behavioral activity was assessed at the end of our chronic, high dose PMO dosing schedule. The results trended toward increased activity in the PMO-treated mice, although not statistically significant. WT mice were not analyzed for each of these parameters (Figures. 23, Supplemental Figures 3, 4, and 5); however, previously published work from our group on functional data and body weights for WT (C57BL/6 mice) [41] compared to mdx can be used as a reference for WT for these assessments. Additional data for WT forelimb and hindlimb strength as shown in Figure. 4 has been previously published by our group, [24]). For example, from this study, mdx forelimb strength is approximately 5 kgF/kg and hindlimb around 8.5 kgF/kg which is similar to what we found for mdx mice in this study. The corresponding WT levels for this are approximately 6.5 and 9.75 kgF/kg for forelimb and hindlimb strength, respectively, demonstrating comparability and that the methods are reproducible. Additionally, we have previously shown that to reach statistical power in pre-clinical behavior analysis, a large sample size (n = 10 minimum) is necessary [21, 42]. In the current study, sample size (n = 8) was limited by the exorbitant cost of the PMO drug, especially at the high dose level. A larger cohort could provide additional data to support statistical significance to increases in activity.
PMO drugs are one of a few shown to restore dystrophin safely in DMD animal models and dystrophic human patients. Our results, in agreement with the literature, show a correlation between dystrophin rescue and force generation in mdx mice [39], indicating that careful measurement of dystrophin protein may become a valid surrogate biomarker to predict functional outcomes in exon skipping. However, we show that the effects of maximal monthly dosing of PMO on function and dystrophin restoration are limited, potentially due to the mechanism by which PMO enters myofibers [35], the half-life of skipped transcript, or turnover of restored dystrophin protein, highlighting the importance of optimizing the dose levels and regimen to achieve reliable success in clinical trials of exon skipping.
Footnotes
ACKNOWLEDGMENTS
Dr. Nagaraju is supported by the NIH (5U54HD053177; K26OD011171, P50AR060836-01; R56NS097229-01), the Muscular Dystrophy Association, and the US Department of Defense (W81XWH-05-1-0616, W81XWH-11-1-0782; W81XWH-11-1-0330) and NICHD (P50AR060836-01 National Center for Medical Rehabilitation Research). We thank Dr. Deborah McClellan for editorial assistance.
CONFLICT OF INTEREST
KN is the co-founder and president of Agada Biosciences and the co-founder and vice-president of research for ReveraGen Biopharma. EPH is the co-founder and vice-president of Agada Biosciences, the co-founder, President and CEO of ReveraGen Biopharma, and the co-founder of TRiNDS. All other authors have no competing financial interests or conflicts of interest to declare.
AUTHOR’S CONTRIBUTIONS
M.B.K., M.C.V., K.N., E.P.H., and J.V.A. designed and supervised the study. M.B.K., M.C.V., J.F.B., K.T., J.Q., J.H.M., J.N., and A.A.F. performed experimental work. U.B. and V.S. performed and analyzed PMO ELISA experiments. M.B.K. and M.C.V. performed primary data analysis. M.B.K., M.C.V, K.N., and E.P.H. wrote the manuscript with significant input from Y.H., T.A.P., Q.L.L., and J.V.A.