
Abstract
Background:
Duchenne Muscular Dystrophy (DMD) is one of the most common muscular dystrophies, caused by mutated forms of the dystrophin gene. Currently, the only treatment available is symptoms management. Novel approximations are trying to treat these patients with gene therapy, namely, using viral vectors. However, these vectors can be recognized by the immune system decreasing their therapeutic activity and making impossible a multidose treatment due to the induction of the humoral immunity following the first dose.
Objective:
Our objective is to demonstrate the feasibility of using a hybrid vector to avoid immune clearance, based on the electrostatic coating of adeno-associated virus (AAVs) vectors with our proprietary polymers.
Methods:
We coated model adeno-associated virus vectors by electrostatic interaction of our cationic poly (beta aminoester) polymers with the viral anionic capsid and characterized biophysical properties. Once the nanoformulations were designed, we studied their in vivo biodistribution by bioluminescence analysis and we finally studied the capacity of the polymers as potential coatings to avoid antibody neutralization.
Results:
We tested two polymer combinations and we demonstrated the need for poly(ethylene glycol) addition to avoid vector aggregation after coating. In vivo biodistribution studies demonstrated that viral particles are located in the liver (short times) and also in muscles (long times), the target organ. However, we did not achieve complete antibody neutralization shielding using this electrostatic coating.
Conclusions:
The null hypothesis stands: although it is feasible to coat viral particles by electrostatic interaction with a proprietary polymer, this strategy is not appropriate for AAVs due to their small size, so other alternatives are required as a novel treatment for DMD patients.
Keywords
INTRODUCTION
Duchenne muscular dystrophy (DMD) is one of the most common forms of muscular dystrophy. DMD is caused by mutations in the X chromosome’s DMD gene that encodes the dystrophin protein. Dystrophin is part of a protein complex that links the cytoskeleton of muscle fibers to the surrounding connective tissue. Without functional dystrophin to support muscle strength and stability, muscle fibers are easily damaged. The dystrophin gene is the largest known human gene, containing 79 exons and spanning more than 2,200 kb. DMD is associated with mutations that disrupt the protein reading frame causing premature stop codons. These mutated transcripts are susceptible to nonsense decomposition, and the carboxy-terminal truncated protein products are also unstable and subject to degradation, leaving little or no protein produced in cells [1, 2].
Therefore, for such a disease caused by alterations in a single gene, a highly appealing therapy is to replace the altered dystrophin with a functional copy using gene therapy. Considering the low stability of gene material in the extracellular environment due to the presence of degradative nucleases, the use of delivery vectors is a must. Among them, viral vectors confer some advantages as compared to non-viral ones. One of the most clinical promising viral vectors for gene therapy are the adeno-associated viral (AAV) vectors.
AAVs are considered a nonpathogenic parvovirus composed of a 4.7 kb single-stranded DNA encapsulated in a non-enveloped capsid (size 20 to 25 nm in diameter). The viral genome is composed of three genes, Rep (essential for site-specific integration), Cap (encodes for structural proteins) and AAP (essential for serotype-specific assembly), flanked by inverted terminal repeats (ITRs) that function as the viral origin of replication and the packaging signal [3]. AAVs infect both dividing and non-dividing cells and are predominantly nonintegrative, which is advantageous in terms of safety issues. The transferred DNA is mostly stabilized as an episome but to a lesser extent, it can remain latent in the host cell DNA by integration into specific chromosomic loci [4]. AAVs require another virus such as adenovirus or herpesvirus to replicate [5].
AAV vectors are developed from a nonpathogenic, nonenveloped parvovirus that is naturally replication-defective. All viral coding sequences in AAV vectors are replaced with a gene expression cassette of interest, fail to integrate in the absence of rep proteins and are able to impart long-term episomal persistence in postmitotic tissues such as muscle [6, 7].
One of the main hurdles in the use of AAV vectors for gene therapy is the host immune system, although AAV are inefficient at transducing antigen-presenting cells (APCs) and have a low immunogenicity profile, exposure to wild-type AAV results in the activation of the immune system against the virus, with development of both humoral and cellular immunity. Furthermore, the majority of the population possesses residual circulating antibodies against AAV due to early exposure in life. As a result, administration of naked AAV vectors elicits a pronounced immune response that gets amplified during the re-administration of the vectors [8].
To solve pre-acquired immunity and re-administration issues, we propose a strategy to make these vectors evade the host immune response during systemic administration that consists in the coating of these viral particles with poly-(beta aminoesters) (pBAEs). pBAEs are biocompatible and biodegradable polymers composed of ester bonds and easily synthesized by one-step Michael addition of primary amines to diacrylates. Different types of pBAEs have been successfully used in several therapeutic applications [9–12]. We have previously showed that oligopeptide end-modified pBAEs (OM-pBAEs) are great non-viral gene delivery vectors with excellent results in terms of transfection efficiency, biocompatibility, and cell specificity in vitro and in vivo [13–15]. In terms of viral therapy, we determined previously that OM-pBAEs have huge potential to coat an oncolytic adenovirus, improving its circulation lifetime, decreasing interactions with neutralizing antibodies, modulating natural tropism as well as the immune response [16].
The main objective of this work is to determine if an electrostatic OM-pBAEs coating of the AAV, as we successfully performed previously with adenoviruses [16], could be a promising alternative as a DMD gene therapy. Specifically, we aim to demonstrate the biodistribution of the viral vectors in muscle tissues as well as the capacity of the coating to prevent from antibody neutralization. We will test two variations of the polymer, including one with poly (ethylene glycol), based on our oncolytic adenoviruses experience.
MATERIALS AND METHODS
Chemicals
All chemical products were purchased from Merck, unless otherwise stated. Reagents and solvents used for synthesis were: CR3 peptide (NH2-Cys-Arg-Arg-Arg-COOH), obtained from Ontores (Shangai) Ltd with a purity of at least 98%.
Cell culture
NSC-34, C2C12 and MOVA cells were obtained from ATCC (Rockville, MD, USA) and cultured following their recommendations. In brief, DMEM-high glucose (Sigma Aldrich) was used as culturing media, supplemented with 10%(v/v) heat inactivated fetal bovine serum (FBS, research grade, HyClone™), 100 units mL–1 penicillin G, 100μg mL–1 streptomycin, and 2 mmol L–1 l-glutamine. Cells were cultured at 37 °C, under a 5%CO2/95%air atmosphere until 90%confluence before starting transductions.
Animals
Adult 6–8 weeks old Balb/c mice were purchased (Envigo) and kept under pathogen-free conditions in laminar flow boxes at controlled environmental conditions of humidity (60%), temperature (22°C±2°C) and light with food and water ad libitum. Animal maintenance and experiments were performed in accordance with established guidelines of the Catalan Government and following protocol number 9938, approved by the Direcció General del Medi Natural.
AAV vectors
Purchased AAV vectors were produced by the Viral Vector Production Unit (UPV) of the Universitat Autònoma de Barcelona. AAV9-GFP (Physical titer: 1,03E + 13 gc/mL) and AAV9-Luciferase (Physical titer: 1,01E + 13 gc/mL) were purified by PEG precipitation / Iodixanol gradient and were stored in PBS-MK / 40%Iodixanol.
METHODS
Preparation of pBAE-coated adeno-associated viruses
Poly(β-amino ester)s (PBAEs) were synthesized as described previously in Dosta et al., and Fornaguera et al., [17, 18]. Specifically, C6-CR3 and C6-CR3PEG polymers were used, at an initial concentration of 100 mg/mL in DMSO [16]. A polymer mixture of 65/35 C6CR3/C6CR3PEG v/v solution was prepared. Then, a physical mixture of the AAV, the polymers (C6CR3 alone or the polymer mixture prepared) and saline at a ratio of 1E-9μg of PBAE / viral particle and a final concentration of 1×1012 VP/mL was prepared in 40%of the final volume in order to facilitate the interaction between the positively charged polymers and the negatively charged virus and incubated for 30 min at 25°C+/-2°, to enable the electrostatic interaction between the negative surface charge of the virus and the positive charge of the polymer. Then, 60%of saline was added by gentle mixing.
Surface charge measurement
Surface charge of nanoparticles were measured at 25°C, 633 nm laser wavelength and 173° signal detector; using a ZetaSizer Nano ZS (Malvern Instruments Ltd, Malvern, UK). Samples were diluted 1/10 in water from size measurements. Three measurements of each nanoparticle batch were performed with 20 runs per measurement, considering the intensity approximation. Results correspond to the mean±standard deviation of at least three independent batches.
Polyacrylamide gel electrophoresis (PAGE)
Polymeric coating was analyzed using the polyacrylamide gel electrophoresis which consists on a gel that separates proteins according to their electrophoretic mobility. Twenty microliters containing 8×1010 VPs of naked AAV and both polymer coatings were loaded onto a 7,5%(w/v) polyacrylamide gel; the samples were electrophoresed at 50 mA for 60 min in 1×PAGE electrophoresis buffer at pH 8.3 (2 M Glycine; 0.25 M Tris-base). Gels were stained with Coomassie Brilliant Blue and washed out with a distaining solution (Glacial acetic acid: methanol: water 1:4:5). Afterwards, gel images were acquired.
Electron microscopy
The morphology, structure and hard-core size of naked and coated viral preparations was determined by transmission electron microscopy (TEM) and field emission scanning electron microscopy (FE-SEM), using Jeol1400 and Hitachi Merlin microscopes. For TEM measurements, samples were first deposited in a carbon-coated copper grid and negatively stained with uranyl acetate prior to imaging. For FE-SEM measurements, a gold coating was performed on the deposited samples. Size analysis was performed using OriginPro software.
In vitro neutralization assay of serums produced in vivo
Balb/C mice were immunized by injecting 4×1011 VP/kg dose of naked AAV9-GFP and C6-CR3/C6-PEG-CR3 (65/35) formulation intravenously at day 0. At day 21 after the injection, blood was collected by puncture of the inferior cava vein. Blood samples were left 30 min at room temperature to induce blood clotting and then centrifuged at 5000 rpm for 15 min. The obtained sera were stored at –20°C.
A total of 1,5 Times; 109 VP/well (MOI: 100.000) were prepared in DMEM with 2%FBS and 25μl of this solution was added to 96-well plates. The serum sample obtained previously was diluted from 1/540 to 1/1,3E5 in DMEM with 2%FBS, and 25μl of each dilution was added to the wells. The mixture was incubated 1h at 37°C. Afterwards, 50μl of the mixture was added to NSC-34 cells (15.000 cells/well) overnight. 24 h post-transduction, 150μl DMEM with 10%FBS was incorporated and the percentage of GFP expression was measured by flow cytometry three days later.
The ND50 values were calculated by determining the dilution which 50%neutralized the signal from the positive transduction control without serum.
In vivo neutralization assay
Balb/C mice were divided in 4 groups and immunized at day 0 by injecting 4 x 1011 VP/kg dose of naked AAV9-GFP (groups 1 and 2, n = 5) or AAV9-GFP coated with C6-CR3/C6-PEG-CR3 (65/35) formulation (groups 3 and 4, n = 5) intravenously. At day 21 after the immunization, mice were injected with AAV9-Luc naked (groups 1 and 3) or coated with C6-CR3/C6-PEG-CR3 (65/35) formulation (groups 2 and 4). We also injected a group (n = 5) with AAV9-GFP naked and AAV9-GFP coated as controls. 6 h after the injection bioluminescent images were taken.
In vivo bioluminescent imaging
Mice were anesthetized i.p. and then injected i.p. with 150μl of luciferin (Regis Technologies) (16.7 mg/ml in saline). Animals were placed in the detection chamber of a high efficiency ImagEM X2 C9100-23BEMCCD Imaging System (Hamamatsu Photonics) and images were acquired from the ventral direction. A second image of the animal was obtained using a white-light source inside the detection chamber, to register the position of the luminescence signal. Light events recorded in images were calculated using the HokawoTM Imaging Software (Hamamatsu Photonics) and expressed as photon counts (PHC) after subtracting the background. The net number of PHCs in the area of interest was calculated using the formula: PHCs = (total number of PHCs in the area of interest) –[(number of pixels in the area of interest)×(average background PHCs per pixel)].
Statistical analysis
Statistical analysis was performed using GraphPad Prism (GraphPad software). Unless otherwise stated, results are given as a mean of three replicates+/s standard deviation. A paired sample t-test was performed to compare means. Differences between groups were considered significant at p values below 0.05 (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
RESULTS
Determination of the pBAE concentration to perform the electrostatic coating
AAVs administered naked in the body are easily recognizable by previously generated neutralizing antibodies, making not possible a second administration and decreasing the effectivity of the treatment if the patient was previously immunized. To avoid these neutralization problems and with the aim to add a targeting moiety in the future, AAVs were coated with poly(beta aminoester) (pBAE) polymers. Specifically, C6CR3 and C6CR3/C6CR3PEG in a 65/35 v/v ratio were used (from now on, they will be named as R-AAV and RPEG-AAV), selected given our previous results of adenoviruses coating [16]. It is mandatory to assess the most appropriate pBAE/AAV ratio, which was performed by measuring the surface charge of the complexes. Figure 1 shows the results of the biophysical characterization as a function of the pBAE/AAV ratio.
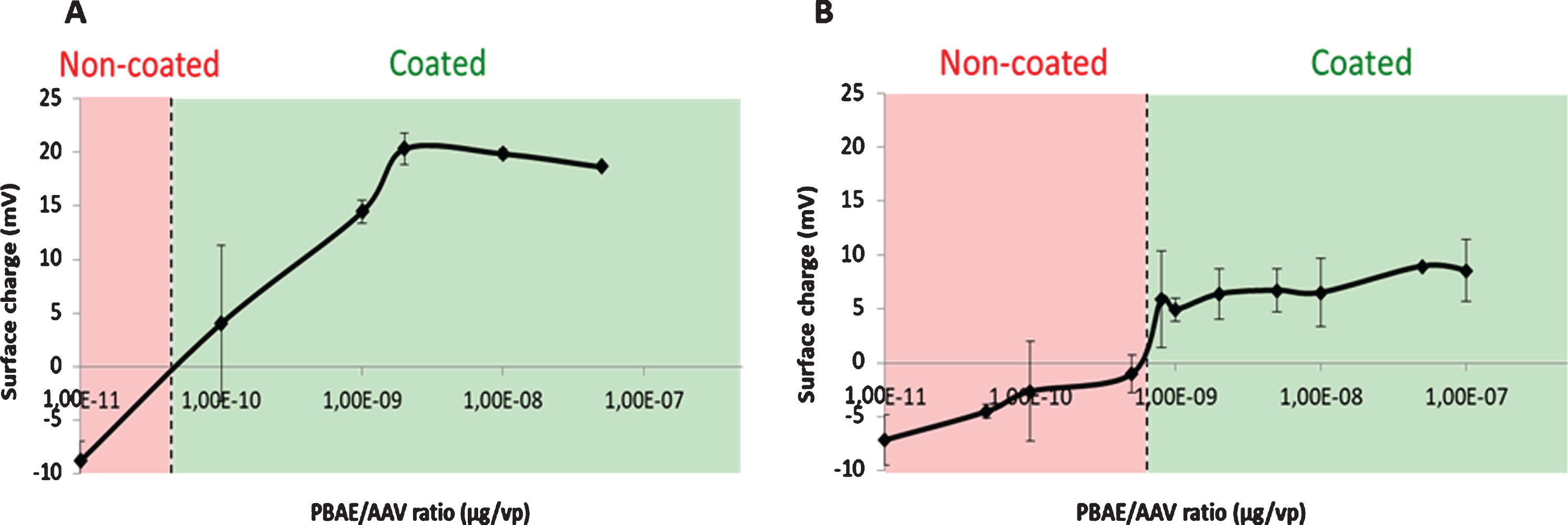
Assessment of the pBAE/AAV coating ratio. Surface charge determination of AAV coated with C6CR3 (A) or C6CR3/C6CR3PEG (B), as a function of the pBAE/AAV ratio.
For both polymers, changes in the surface charge of coated particles reached a plateau at 1E-9μg of polymer/viral particles and this result is confirmed with polyacrylamide gel electrophoresis assays (Figure S1), so this ratio was selected to continue with the viral coating characterization.
Generation and biophysical characterization of pBAE-coated AAVs
Once selected the pBAE/AAV ratio, naked and coated AAV sizes and morphology were characterized by electron microscopy. First, TEM was proposed as the technique of election, based on our previous experience on adenoviruses coating [19]. However, as it can be seen in Fig. 2, only a small number of AAV could be found by this technique, so we also included SEM technique for the analysis of the morphology and size of AAV coating. As it can be seen in Figs. 2 3, naked viral particles showed sphere shape, as expected, and the coating can be confirmed again thanks to an increase from the 20 –25 nm of initial diameter of AAV to the > 35 nm polymer coating. While for TEM size increase was slight and very similar when using both polymers, by SEM technique, sizes showed a higher increase, up to around 50 / 60 nm and showed significant differences between R-AAV and RPEG-AAV coating. To remark, the presence of aggregates in the R-AAV coating, of sizes bigger than 135 nm, make that the use of R-AAV coating is not as efficient as that of RPEG-AAV coating. Nevertheless, being all sizes below 200 nm, we proceeded with both coatings for further studies.
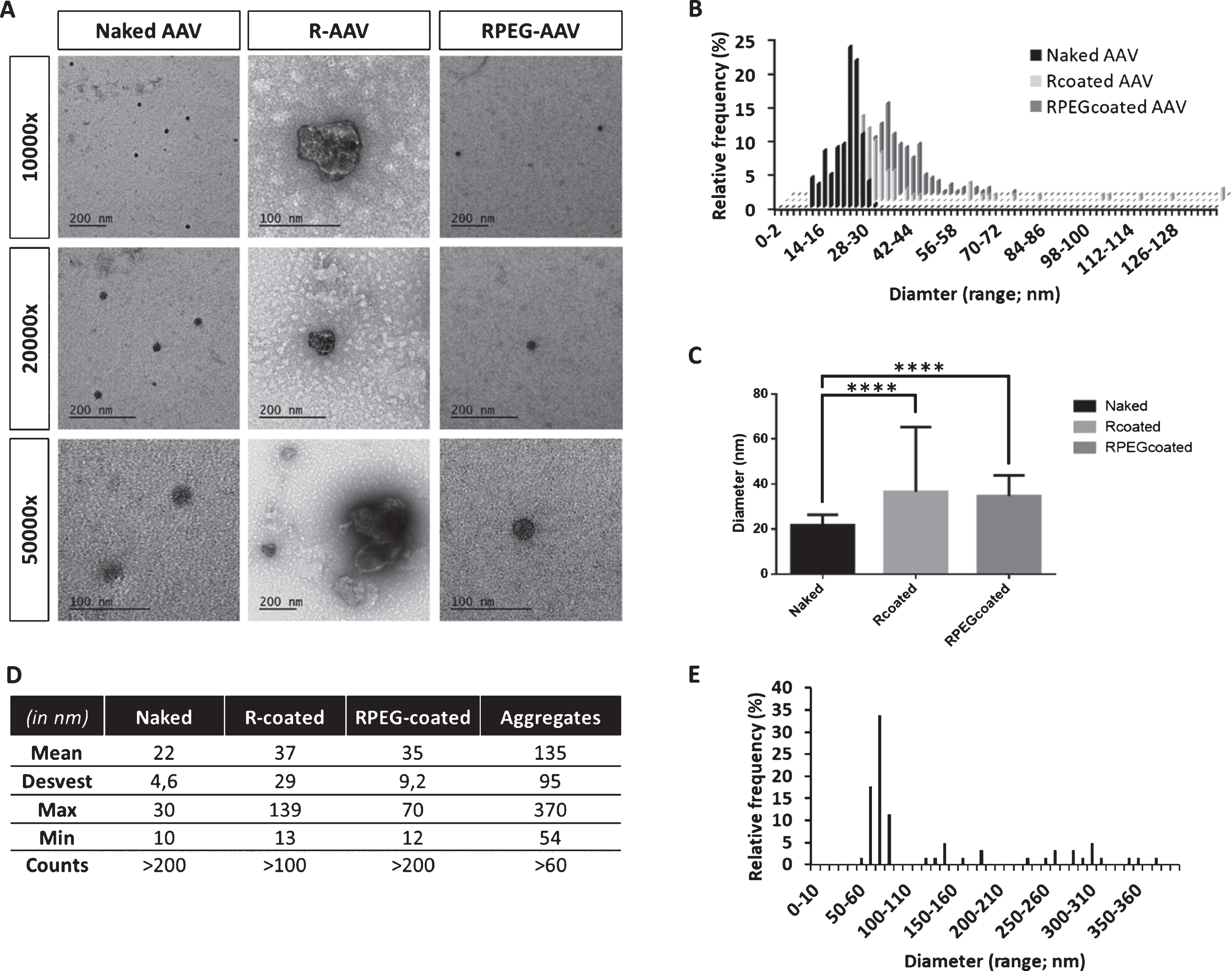
TEM analysis of naked and coated AAVs. A –Micrographies at different magnifications; B - Size distribution of AAVs diameters; C –Statistical analysis of the mean diameter comparation; D –Summary table of the statistical analysis performed; and E –Size distribution of aggregates in R-coated AAVs. ****p < 0.0001.
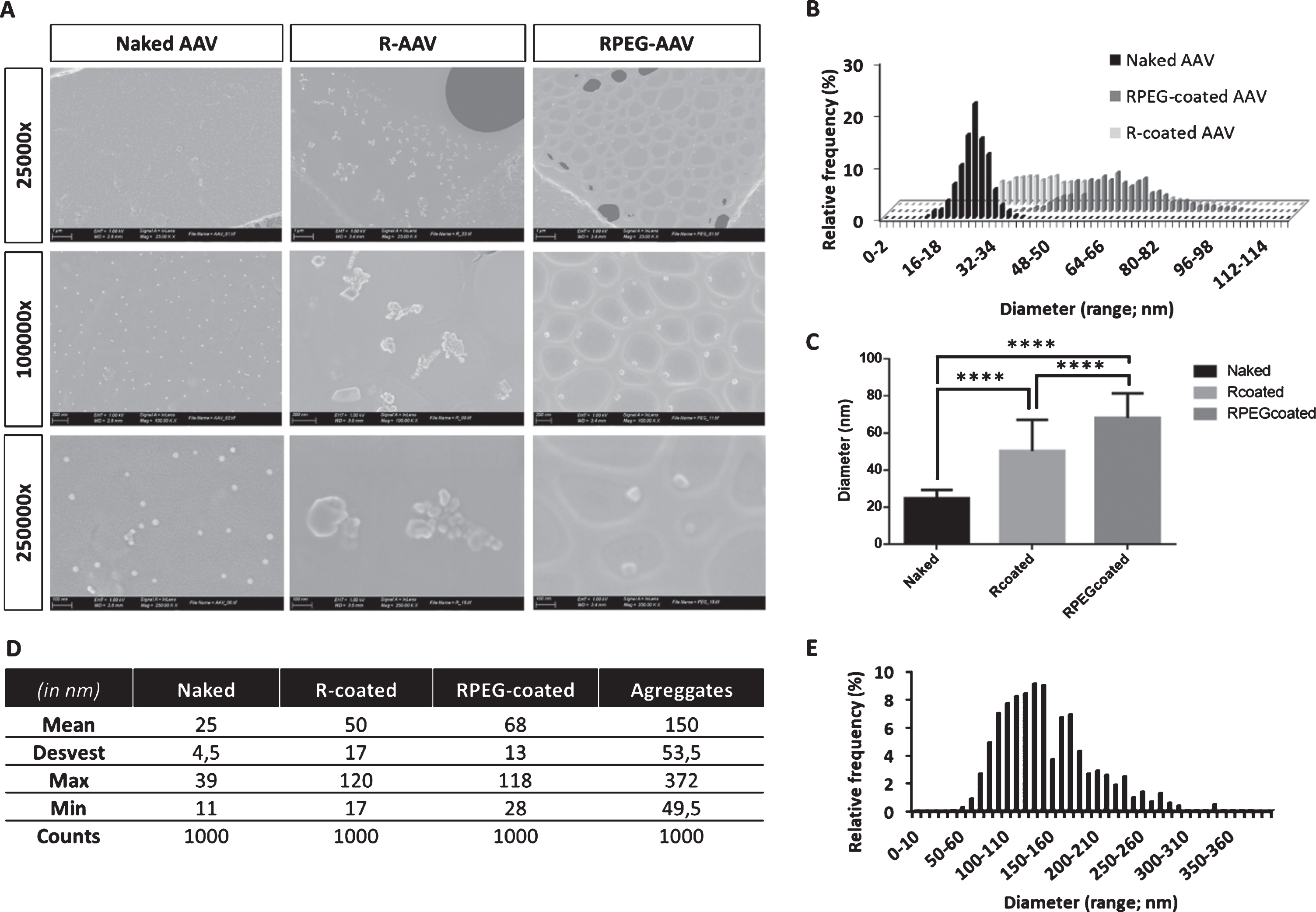
SEM analysis of naked and coated AAVs. A –Micrographies at different magnifications; B - Size distribution of AAVs diameters; C –Statistical analysis of the mean diameter comparation; D –Summary table of the statistical analysis performed; and E –Size distribution of aggregates in R-coated AAVs. ****p < 0.0001.
In vivo biodistribution studies suggest that AAV expression could be attributed to muscle tissue accumulation
Further, we proceeded with biodistribution studies to determine if AAVs, once coated, were able to transduce preferentially muscle tissues. 4×1011 VP/kg of naked and C6-CR3/C6-PEG-CR3 (65/35) coated AAV9-Luciferase were injected in Balb/C mice and as shown in Fig. 4, after a very short period (7 days), AAVs seem to transduce only the abdominal region, which could be attributed to liver accumulation. However, after longer times (from 14 days), the signal spreads through the whole body of the animal, which could be an indication of the muscle transduction preference. In fact, the maximum expression was found between 14 / 28 days, specifically in the legs of the mice, which agrees with the idea of muscle tissue transduction. Quantification of the muscle bioluminescence - BLI (Fig. 4B) resulted in a slight increase in the bioluminescence with the coated viral particles. Further, we analyzed the muscle / liver BLI ratio, as shown in Figure S2, only in RPEG-coated 8E-10 AAV an increase on this ratio was observed, thus confirming this formulation is the most appropriate for further studies.
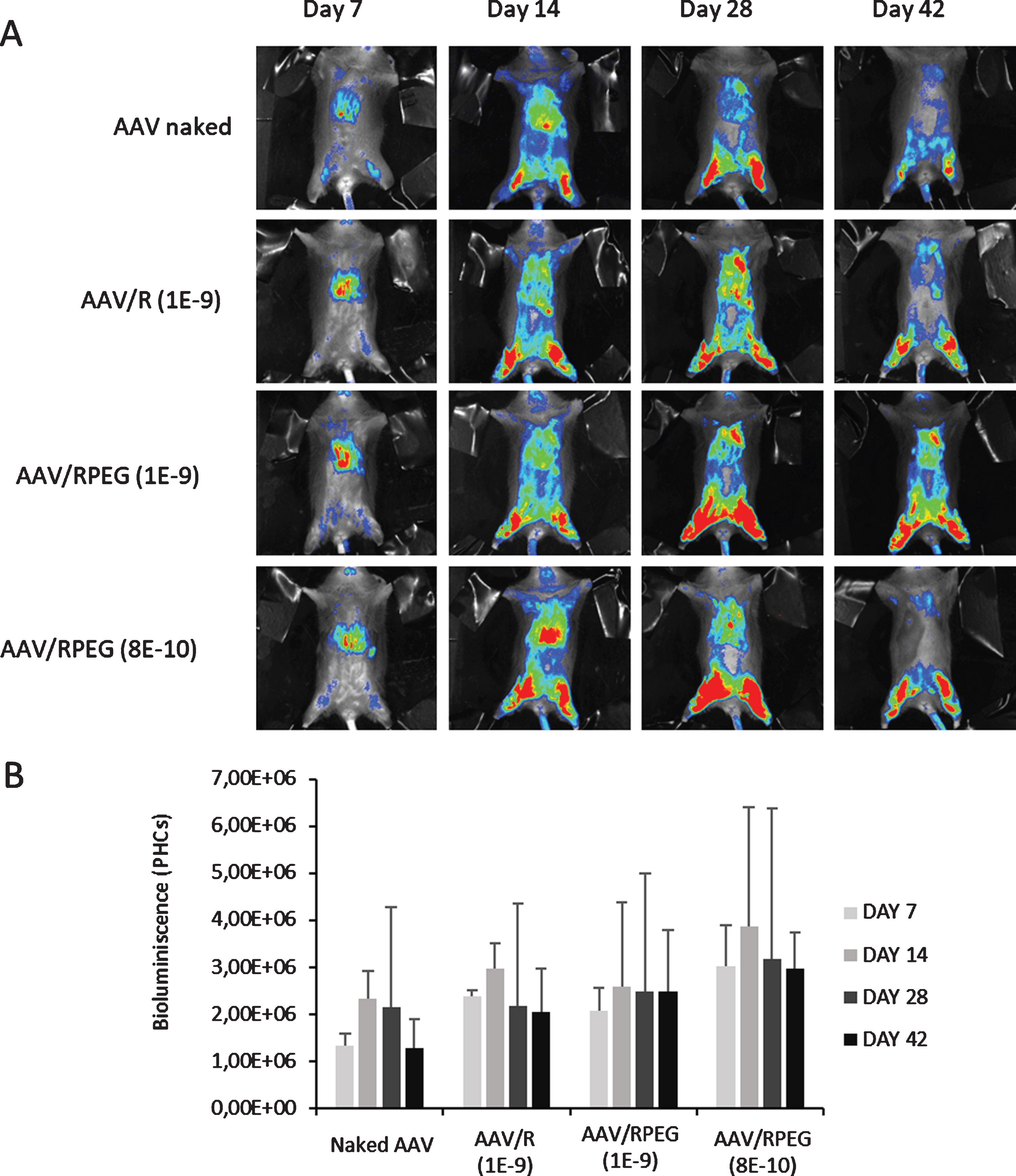
In vivo biodistribution studies. 4×1011 VP/kg of AAV9-Luciferase naked or coated were injected to Balb/C mice and the expression of the transgene were quantify by Bioluminescence images. A –Bioluminiscence images of a representative mouse per group; B –Quantified muscle bioluminescence of each group, as a function of the treatment administered and time; Data points show the average of triplicate measurements; bars, standard deviation of the mean. No significant differences were found between groups.
Therefore, in the following, the RPEG coating was selected and compared with naked AAVs.
Mice injected with RPEG electrostatic coated AAVs produce neutralizing antibodies
To obtain serum with neutralizing antibodies, 8 Balb/c mice were injected with AAV9-GFP naked (n = 4) or AAV9-GFP coated with RPEG at a ratio of 65/35 (n = 4). In this experiment, the R-coated AAV wasn’t studied since, as aforementioned, the RPEG-coated 8E-10 AAV was the only formulation that presented a higher muscle/liver BLI ratio compared to the other samples. 3 weeks after the inoculation, a second injection with the same groups were done. One week after the second injection, serum was purified and 1,5 x 109 VP/well (MOI: 100.000) of AAV9-GFP were incubated with different dilutions of the extracted sera. After the incubation, transduction of NSC-34 cells was analyzed by flow cytometry and the titer of the neutralizing antibodies was calculated (Fig. 5A). There were no significant differences between the naked and the coated inoculation, meaning that the same amount of neutralizing antibodies was produced. In parallel with these in vitro experiments, an in vivo study of the neutralizing antibodies production was performed. Balb/C mice were divided into 5 groups, 3 of these groups received a first inoculation with AAV9-GFP naked (2 groups) and AAV9-GFP/RPEG (1 group). 3 weeks after the inoculation, a second injection of AAV9-Luciferase naked or coated were administered following the scheme in Fig. 5C. There was no luciferase activity in any of the 3 groups pre-immunized with either AAV9-GFP nor AAV9-GFP/RPEG while a detectable amount of light was present in groups 4 and 5, where no preimmunization was produced. These results, together with the in vitro experiments, confirm the production of neutralizing antibodies when mice were injected with coated AAV, considering that no expression or activity of the luciferase gene was observed.
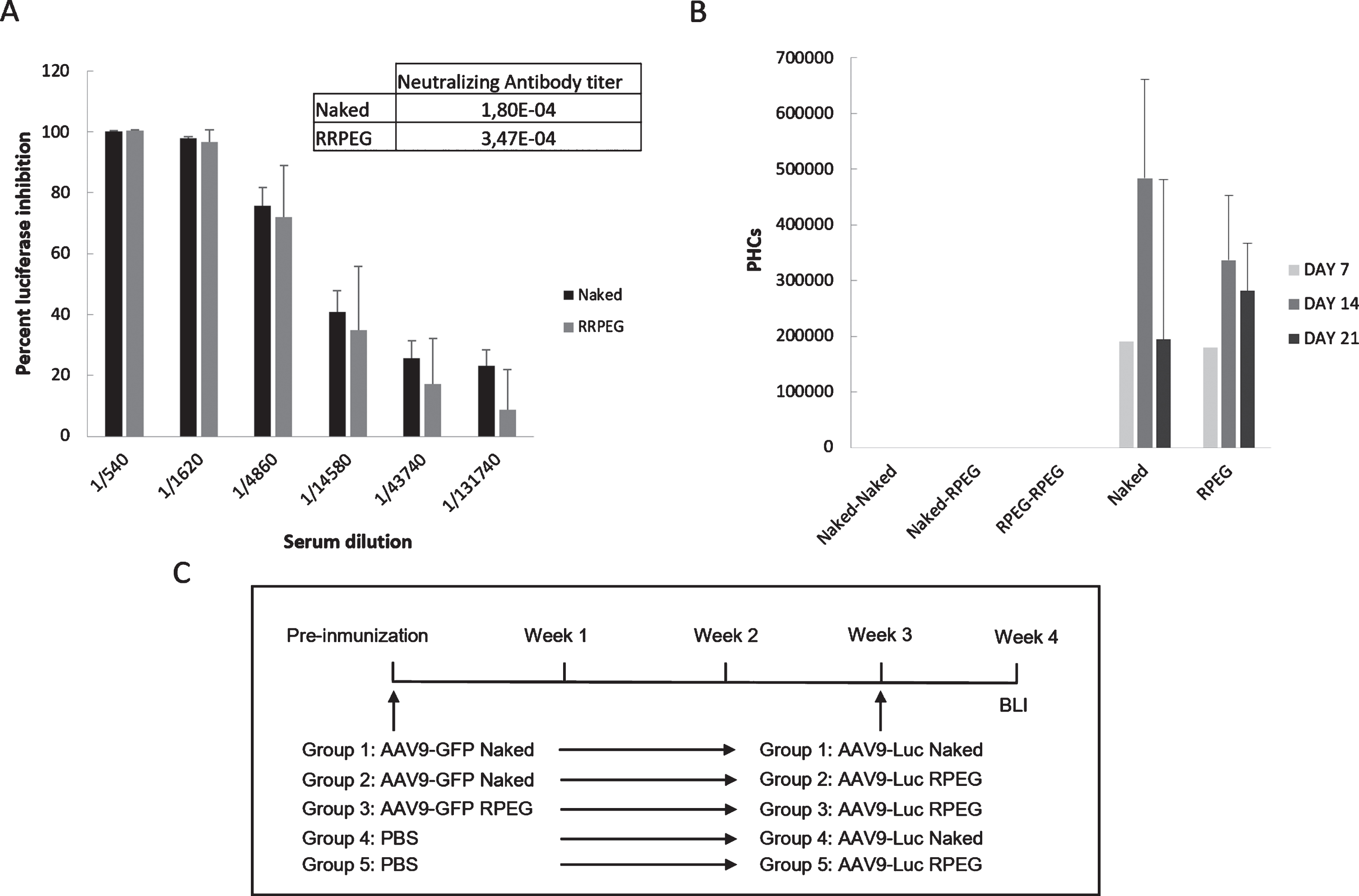
Neutralizing antibodies capacity of coated AAVs. A –Percent of luciferase expression inhibition with different dilutions of serum from AAV naked/coated injected mice, n = 5 per group; and B –Photon counts quantified after preimmunization with AAV9-GFP naked or coated with RPEG in mice. N = 5 per group C –Schematic representation of the in vivo antibody neutralization assay where the different experimental groups were defined.
DISCUSSION
Duchenne Muscular Dystrophy (DMD) is one of the most common muscular dystrophies caused by genetic congenital mutations consisting on the lack of a functional copy of the dystrophin gene, required to maintain the structure of muscle fibers. Currently, there are no efficient therapies and children born with this condition die before they reach adulthood. Although much effort has been devoted in the field of gene therapy to the replacement of the non-functional dystrophin copy by the use of viral vectors, these are recognized by the patient’s immune system and are removed from circulation before they perform their therapeutic action. Therefore, there is still an urgent need to evade the immune system’s recognition [20].
In this context, we proposed the use of coated adeno-associated vectors (AAVs), based on our previous experience with adenoviruses coating [19], and on the available bibliography on the field [21, 22], to shield AAVs from the host immune system. In our previous study [16], we successfully achieved shielding from antibody neutralization by coating an adeno-associated vector with our proprietary polymers, poly (beta aminoesters) –pBAEs, including a sample with a poly-(ethylene glycol) –PEG moiety on the surface. In here, we followed the same principle to study if the electrostatic coating strategy, successful for the coating of adenoviruses, could also work for the coating of much smaller viral particles: adeno-associated viruses. Although being aware of the controversy over the use of PEG due to the development of some immunogenic reactions reported in previous studies [23–25], it remains the gold standard for promoting the stealthy surface of the resulting coated AAVs [26–28]. Therefore, we tested two coatings, with and without PEG moieties.
The coating was formed by the electrostatic interaction between the anionic viral surface and the cationic polymers, achieved by the specific use of arginine-modified pBAEs, as previously demonstrated when complexing different types of nucleic acids and adenovirus coatings [14, 29]. It was first determined that we needed, at least, 1E-9 ug pBAE / viral particles to achieve the complete coating of the capsid, as assessed by a shift from the anionic to the cationic surface (Fig. 1). Once this ratio was known, to confirm that the coating was effectively produced, we performed electron microscopy experiments (Figs. 2 3). We clearly observed that, although the spherical shape of the naked AAVs was somehow maintained (some more elongated particles were observed), the sizes increased significantly when we coated the AAVs. pBAE-PEG-coated AAVs remained individually in the sample, with sizes around 50 nm. These sizes are much larger than those reported for naked AAV, so this is confirmation that the coating took place. In addition, the use of pBAE without the PEG moiety produced aggregates in the samples, of around 150 nm, which could be attributed to the inclusion of more than one AAV in a single coated unit. These sizes are compatible with parenteral administration, although the suspected aggregation of several AAV particles in a single polymer aggregate is not desired for further use, mainly due to the lack of control of their behavior once administered. Therefore, although being aware of this problem of the aggregates, since some individual coated particles were also observed, we decided to proceed with both coatings and compare them in further studies.
Once the structural integrity of the coated AAVs was confirmed, we performed functional studies directly on mouse models, using AAVs that codified for the luciferase enzyme, to easily detect the organs in which the AAVs were able to transduce. It is worthy to mention that doses used in this experiments, based in our previous experience (confidential results) were lower than most existing bibliography, although in some articles, they had already used similar doses of AAVs [30]. In fact, the low doses represent a competitive advantage in terms of reducing production costs and avoiding putative side effects that may be generated from AAV accumulation. AAV9 has a natural tropism to liver, muscle, heart and CNS [31]. At early times we mainly observed the liver localization, as it can be clearly seen in the different images of day 7, and as the experiment progressed the BLI signal was also observed in the legs of the mice (Fig. 4). Aiming to develop a gene therapy for DMD patients, we envisage a muscular accumulation, instead of liver transduction, so we quantify the muscle/liver ratio expecting an increase in the ratio when the AAV particles were coated. Our results show the highest ratio when the polymer with PEG was used, indicating that PEG is required to increase the stability of the coating and to tune the biodistribution to preferential transduction of muscle cells, as compared to liver accumulation. In previous studies, we have already reported the modulation of the biodistribution of nanosystems, after parenteral administration, by tailoring our pBAE proprietary polymers. Even without the addition of active targeting moieties, we have demonstrated that by playing with the terminal end-oligopeptide combination included in our pBAE polymers, we could shift the liver accumulation to other organs [14, 29]. However, in this specific case, the coating with the C6CR3-pBAE did not achieve the entire liver avoidance. These results can be attributed to the presence of aggregates in the coatings without PEG, which could hinder the escape from the reticuloendothelial system premature clearance, which takes place after liver accumulation by phagocytosis of the nanosystems. Nevertheless, as suspected by electron microscopy studies, the addition of PEG to the pBAE structure is key to achieving this de-localization.
Although these results were promising, when we further studied the ability of the C6-CR3PEG coating to prevent antibody neutralization, the results were disappointing.
We assessed the capacity of our coating to decrease the amount of neutralizing antibodies using two different approaches. In the first one, we extracted the serum from pre-immunized mice, incubated it with naked AAV particles and then measured the transduction capacity of these viral particles and calculated the neutralizing antibody titer. In the second one, the mice were pre-immunized with AAV9-GFP, then inoculated with AAV9-Luciferase naked or coated and the bioluminescence signal was measured to demonstrate the presence of neutralizing antibodies against the viral particles. Although a shielding from antibody neutralization was expected as extrapolated from adenoviruses coating, in both cases here, there is no difference between the naked or coated particles, meaning that the coating is not sufficiently stable when injected intravenously and that neutralizing antibodies are present in the sera. This lack of stability was attributed to a smaller size of AAVs as compared to adenoviruses, which makes that their surface charge density is not enough to function as a nucleation point to maintain the coating in physiological conditions, where there are a lot of proteins competing for electrostatic binding.
In conclusion, although structural techniques confirmed the formation of an electrostatic coating on AAVs, and this coating produced a shift of the biodistribution to an enhanced accumulation in muscle cells, the interaction between the polymer and the AAV is not stable enough in physiological conditions to protect the AAVs from antibody neutralization.
To sum up, as briefly mentioned above, we have previously demonstrated that pBAEs are able to stably coat viral particles (an oncolytic adenovirus vector) increasing the lifetime of the particles once were injected intravenously and decreasing the recognition by neutralizing antibodies [19]. The main difference with AAV vectors is the size of the particles, while AAV are very small (20–30nm), adenovirus measure between 90 and 100 nm. These differences could make that the charge density of the nanoparticle, once coated with the pBAES, was not enough to maintain the electrostatic coating, making the electrostatic AAV covering non-stable once it is injected intravenously.
In the future, we will modify our proprietary polymers to obtain a stronger interaction and achieve a more stable coating of the AAV vectors or, alternatively, we could use adenoviruses for muscular dystrophies gene therapy. Stronger coatings were tested by other authors with other polymers and for other applications [21, 32–35] but they only showing a moderate protection from antibody neutralization; therefore, there is a long way to go to discover a novel DMD treatment based on polymeric coated AAVs.
Footnotes
ACKNOWLEDGMENTS
Financial support from MINECO/FEDER (grant RTI2018-094734-B-C22) is acknowledged. The Support of Agència de Gestió d’Ajuts Universitaris i de Recerca (AGAUR) from Generalitat de Catalunya for their support trough SGR 2017 1559 grant is acknowledged. Duchenne Parent Project España (DPPE) is also acknowledged for their financial support. Authors acknowledge Elena García and Irene Porcar for their kind support in polymer synthesis and some experiments performance.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.