
Abstract
Background:
Duchenne muscular dystrophy (DMD) is an X-linked disorder caused due to large deletions, duplications,and small pathogenic variants. This article compares the carrier frequency of different pathogenic variants in the DMD gene for the first time in an Indian cohort.
Methods:
Ninety-one mothers of genetically confirmed DMD probands are included in this study. Pathogenic variants in the DMD gene in probands were detected by multiplex ligation-dependent probe amplification (MLPA) or next-generation sequencing (NGS). Maternal blood samples were evaluated either by MLPA or Sanger sequencing. The demographic and clinical details for screening of muscle weakness and cardiomyopathy were collected from the confirmed carriers.
Results:
Out of 91 probands, large deletions and duplications were identified in 46 and 6 respectively, while 39 had small variants. Among the small variants, substitutions predicted to cause nonsense mutations were the most common (61.5%), followed by frameshift causing small insertion/deletions (25.6%) and splice affecting intronic variants (12.8%). Notably, 19 novel small variants predicted to be disease-causing were identified. Of the 91 mothers, 53 (58.7%) were confirmed to be carriers. Exonic deletions had a significantly lower carrier frequency of 47.8% as compared to small variants (64.1%). The mean age of the carriers at evaluation was 30 years. Among the carriers, two were symptomatic with onset in the 4th decade, manifesting with progressive proximal muscle weakness and dilated cardiomyopathy.
Conclusion:
Carrier frequency of small pathogenic variants differs significantly from large deletions. Small pathogenic variants are more commonly inherited, whereas large deletions arise de novo.
Keywords
INTRODUCTION
Duchenne muscular dystrophy (DMD) is the most common inherited childhood muscle disease with a pooled global birth prevalence of 19.8 per 100,000 live male births [1]. It is an X-linked recessive disorder caused due to pathogenic variants occurring in the DMD gene (Xp21.2), which is the largest known human gene spanning 2.4 Mb and comprising of 79 exons [2]. Depending on the reading frame changes the pathogenic variants can result in DMD or Becker muscular dystrophy (BMD) [3]. Pathogenic variants in the DMD gene include large exon deletions, duplications, single nucleotide variants (SNVs) and small insertions/deletions/insertion-deletion variants (together addressed as small pathogenic variants). Large deletions and duplications are the most common pathogenic variants (70– 73%) followed by small pathogenic variants (25– 30%) [4, 5].
Determining the carrier status of the mothers of children diagnosed with DMD is vital for genetic counseling, family planning, and prenatal diagnosis. Though the inheritance is X-linked and mothers are expected to be obligate carriers, nearly one-third of the males with DMD may not have any family history and were often found to have a de novo pathogenic variant [6]. Among the female carriers, around 2.5– 10% may be symptomatic with muscle and cardiac symptoms, and this could vary from mild to severe manifestations [7, 8]. Although there are several studies on the natural history, phenotype - genotype correlation and mutational patterns in DMD patients from India; studies on carrier frequency and inheritance rate of various types of pathogenic variants are few [9–13]. In the current study, we have tested a large number of mothers of DMD probands for carrier status and determined the carrier frequency for the various pathogenic variants.
METHODS
Study participants and protocol
This retrospective observational study involved 91 mothers of genetically confirmed DMD children who visited the neuromuscular clinic of a quaternary hospital in southern India between 2015 and 2020. Institutional ethics committee (IEC) approval (NIMHANS/DO/103RDIEC/2016), informed consent from patients and families were obtained for this study. For the convenience of representation in this study, the term ‘proband’ refers to genetically confirmed DMD patients and ‘carrier’ refers to mothers established as positive for DMD mutation identified in the proband. DMD was diagnosed based on clinical findings, serum creatine kinase (CK) level, and genetic analysis. Multiplex Ligation-Dependent Probe Amplification (MLPA) and Next-Generation Sequencing (NGS) testing were undertaken for suspectedcases of DMD as performed previously by Polavarapuetal., from the same institute [5]. Thesequence of evaluation of mothers for carrier status is depicted in Fig. 1. Blood samples were collected from the mothers for genetic analysis to confirm carrier status after written informed consent. Genomic DNA was extracted using a Qiagen bloodmini-kit. MLPA was carried out if the proband’s variant was a large deletion or duplication, whereas Sanger sequencing was performed if probands had small pathogenic variants.
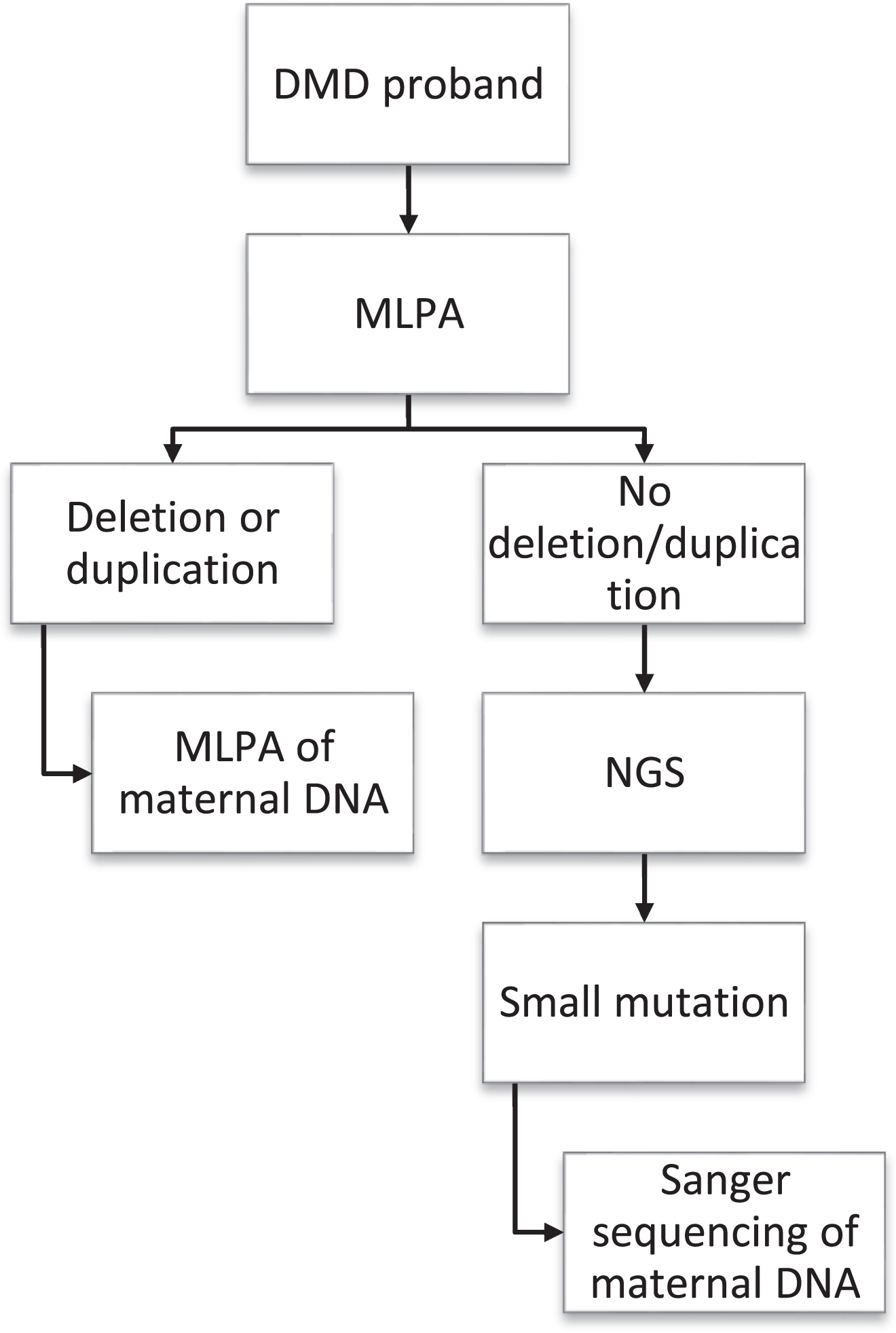
Flowchart depicting the sequence of evaluation of carriers in the study.
MLPA
MLPA was performed using SALSA MLPA kit (MRC-Holland) with probe sets P034 and P035 for the 79 exons of DMD gene as per manufacturer’s protocol. The fragment analysis was performed in a 3500XL capillary electrophoretic machine with LIZ600 as a size standard. For each batch, along with test samples, 2-3 negative controls (DNA negative for large deletions or large duplications) and one positive control were run. The data was analyzed using Coffalyser.net (MLPA analysis software MRC-Holland).
Sequencing
For Sanger sequencing, the primers were first designed for each of the exons. Depending on the pathogenic variant, the respective primers were selected for the mothers. Polymerase chain reaction (PCR) amplification was performed. After PCR clean up with ExoSap-IT, cycle sequencing was performed using BigDye Terminator v3.1 cycle sequencing kit as per the manufacturer’s protocol. Post cycle sequencing, a round of clean-up was performed using the NucleoSeq column method. The cleaned-up fragments were run in a 3500XL capillary electrophoretic machine, and the electropherogram was analyzed. Each nucleotide change was verified by reverse sequencing.
Interpretation of sequence variants
Variants were described with reference to the GenBank transcript, NM_004006.3. All variants identified were compared with the available data in Clinvar, Human Gene Mutation Database (HGMD), DMD gene variant database (https://www.LOVD.nl/DMD) or reported variants in the literature in context of DMD. The variations not reported were considered as novel. We analysed small variants and exonic deletions/duplications in https://www.dmd.nl/DOVE/ for variant annotation and predicted consequence. Classification of pathogenicity is according to 2015 American College of Medical Genetics and Genomics (ACMG) criteria [14]. We used free version of VarSome online tool (https://www.varsome.com/) to automatically generate predictions for small variants on the different ACMG criteria except PS2, PS4, PM6, PP1, PP4, BP2, and BP5. Further, additional criteria, if required, were manually specified based on the variants’ information. PP4 was considered for all variants, and PS2 criteria was included for cases where the mother did not harbor the variant identified in the proband.
Demographic and clinical data
Demographic data and clinical history to screen for muscle and heart involvement, including progressive limb weakness, altered gait, muscle hypertrophy, cramps, myalgia, exercise intolerance, and cardiac symptoms, were collected from the confirmed carriers through telephonic and video interviews.
Statistical analysis
Descriptive statistical analysis was carried out using IBM SPSS Version 26 employing the Fisher’s exact test or the Chi-square test as appropriate. The differences were deemed statistically significant at a P-value < 0.05.
RESULTS
The pattern of disease-causing variants in probands
Among the predicted pathogenic variants in the probands, there were 46 large deletions (50.5%), 6 large duplications (6.6%), and 39 small variants (42.9 %). Among the large deletions, all except one were predicted to be out-of-frame pathogenic variants. Large deletions involving the hotspot region (exon 45– 54) were present in 39 (84.8%), while 11 (23.9%) had single exon deletion. The most common multi-exon deletion involved 45– 50 exons, and single exon deletion frequently affected exon 45. The large duplications noted were exon 2– 7, 3– 7, 21– 30, 52– 76, 56– 74, and 61– 70. Out of the 39 patients with small variants, single nucleotide substitutions accounted for 29 (74.3%) of them and among these, 24 were predicted to result in nonsense pathogenic variants, while 5 were identified as intronic splice affecting variants either involving canonical splice site regions (2) or splice proximal regions (3). In 10 probands, small deletions and/or insertions predicted to cause a frameshift and truncated protein were identified. Notably, 19 (48.7%) small variants reported in this study are novel, while others have been previously reported in DMD patients (Table 1 and Supplementary Table 1). Family history of DMD (with the involvement of 2 or more family members) was present in 31 (33.7%) children, whereas 57 (61.9%) were sporadic cases and details unavailable for 4 cases.
Summary of 53 carrier positive families with description of variants and clinical phenotype of the carriers
*In silico based predicted consequence at protein level. ∧Predicted In-frame deletion/duplication. SNV: Single nucleotide variation/substitution; Del: Deletion; Dup: Duplication; Ins: Insertion; ACMG: American college of medical genetics. Note: Mutations described in this study were submitted to LOVD database. Database Ref: ‘Nalini 2021, submitted’.
Carrier frequency
Of the 91 mothers tested, 53 were carriers with an overall carrier frequency of 58.2%. Among the positive carriers, 25 (47.2%) had small variants, while 28 (52.8%) were positive for large deletions and duplications. No mutation was detected in 38 of the remaining mothers. In four families, with apparent X-linked recessive family history (≥2 affected males), mothers were negative for DMD mutations identified in probands.
The difference among sporadic and familial cases
Among the mothers who had significant family history with 2 or more family members involved, 27 of the 31 (87.1%) tested were detected to be carriers. In comparison, 24 of the 57 mothers (42.1%) from sporadic families were carriers (Fig. 2). The carrier frequency in familial cases was significantly higher than sporadic cases (P-value < 0.001).
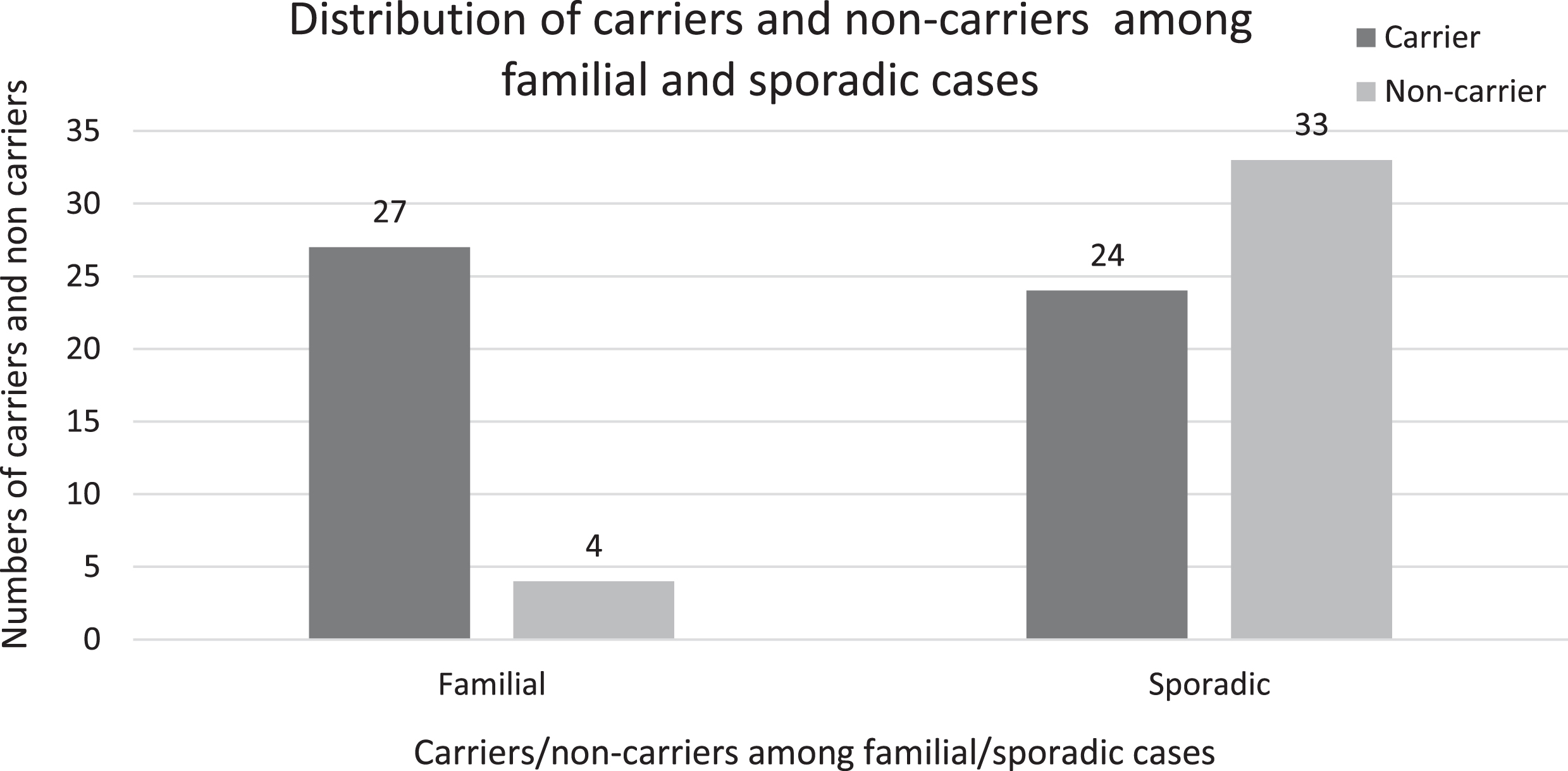
Bar diagram depicting the distribution of carriers and non-carriers among the familial and sporadic cases. Carrier frequency in familial cases was significantly higher than sporadic cases (P-value < 0.001).
Clinical details of carriers
The mean age of the carriers at evaluation time was 30 years (range: 26 to 45). Among the 53 confirmed carriers contacted, 23 mothers responded to the follow-up telephone interview. Eleven mothers had clinical symptoms such as cramps, myalgia, easy fatigability, and exercise intolerance. Two mothers were definite symptomatic carriers with significant progressive lower limb weakness with positive Gower’s sign and calf hypertrophy. One was aged 40 years with onset of symptoms at the age of 35, while the other was aged 42 years and had symptom onset at age 34. Among the mothers with lower limb weakness and significant cardiac symptoms, elevated serum CK (554 U/L) and evidence of cardiomyopathy with dilated left ventricle were confirmed by echocardiography in one mother. The pathogenic variants noted in symptomatic carriers were 45– 47 exon deletion and a susbstitution causing nonsense mutation c.8530C > T (p.Gln2844Ter) in exon 57.
Carrier frequency as per the type of pathogenic variants
Out of the 46 mothers whose sons had large deletions, 22 were confirmed to be carriers (47.8%) (Table 1). Carrier frequencies for duplication and small pathogenic variants were 100% (6/6) and 64% (25/39), respectively (Fig. 3). A significant association was found between the carrier frequencies and the type of pathogenic variant (P-value = 0.03). In particular, the difference in the carrier frequencies was found to be significant between deletional (47.8%) and non-deletional pathogenic variants (68.8%) analyzed (P-value = 0.028). Carrier frequencies for hotspot and non-hotspot large deletions were 51.2% (20/39) and 28.5% (2/7) respectively, with no significant difference in the rate of inheritance (P-value = 0.268). No significant difference was observed between the carrier frequency of single exon large deletions (45.5%) and multiple exon large deletions (48.5%).
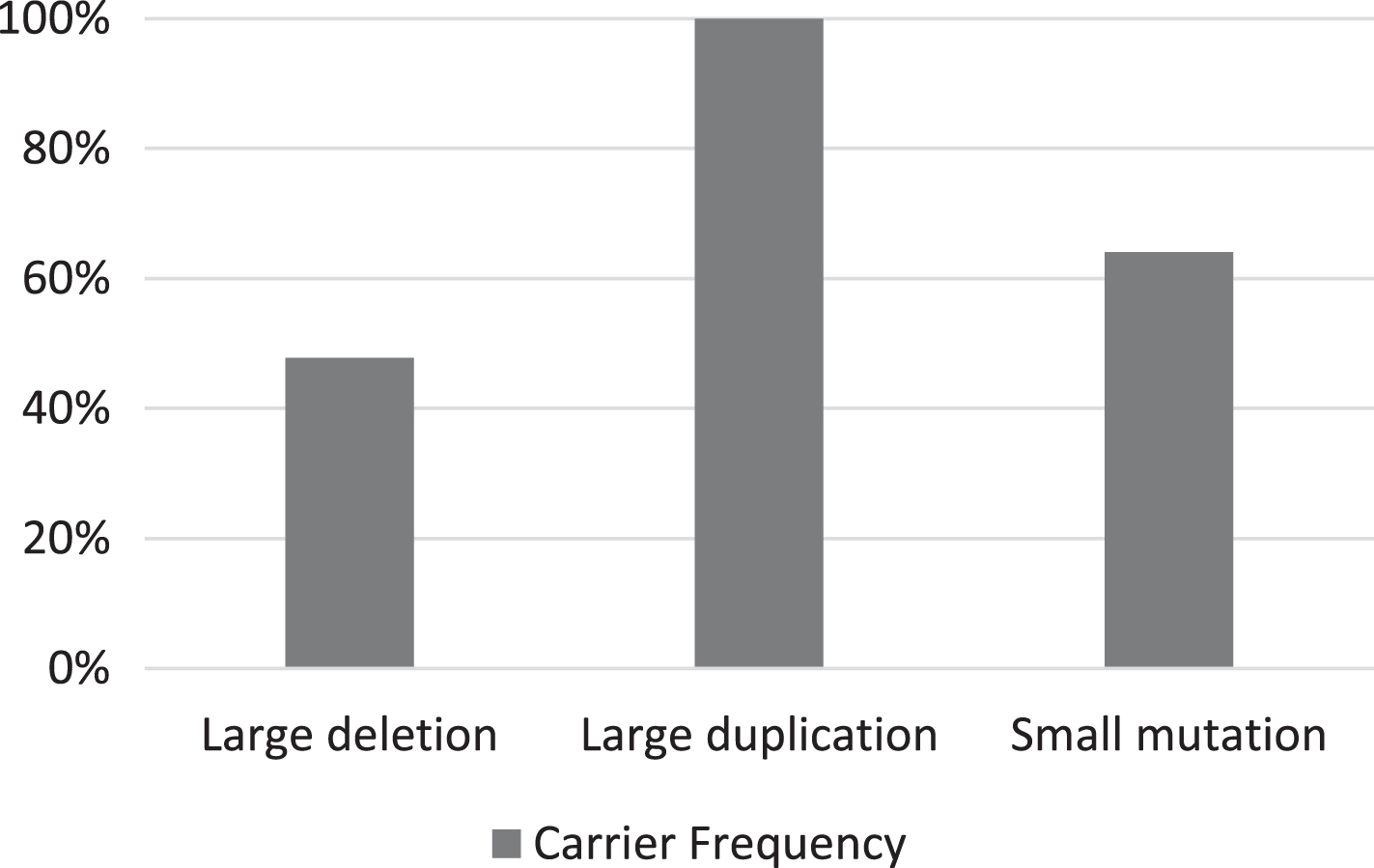
Bar diagram representing the carrier frequencies for large deletion, large duplication, and small pathogenic variants. The carrier frequency for large deletions was significantly lower than the other pathogenic variants (P-value = 0.03).
Among the different types of small variants, the carrier frequency was 80% (8/10) for frameshift, 66.6% (16/24) for nonsense and 20% (1/5) for intronic splice region variants (Fig. 4). However, the difference in the carrier frequency among the small variants was not significant (P-value = 0.054), although there is a trend. The small pathogenic variants confirmed by Sanger sequencing in the carriers are shown in Table 1.
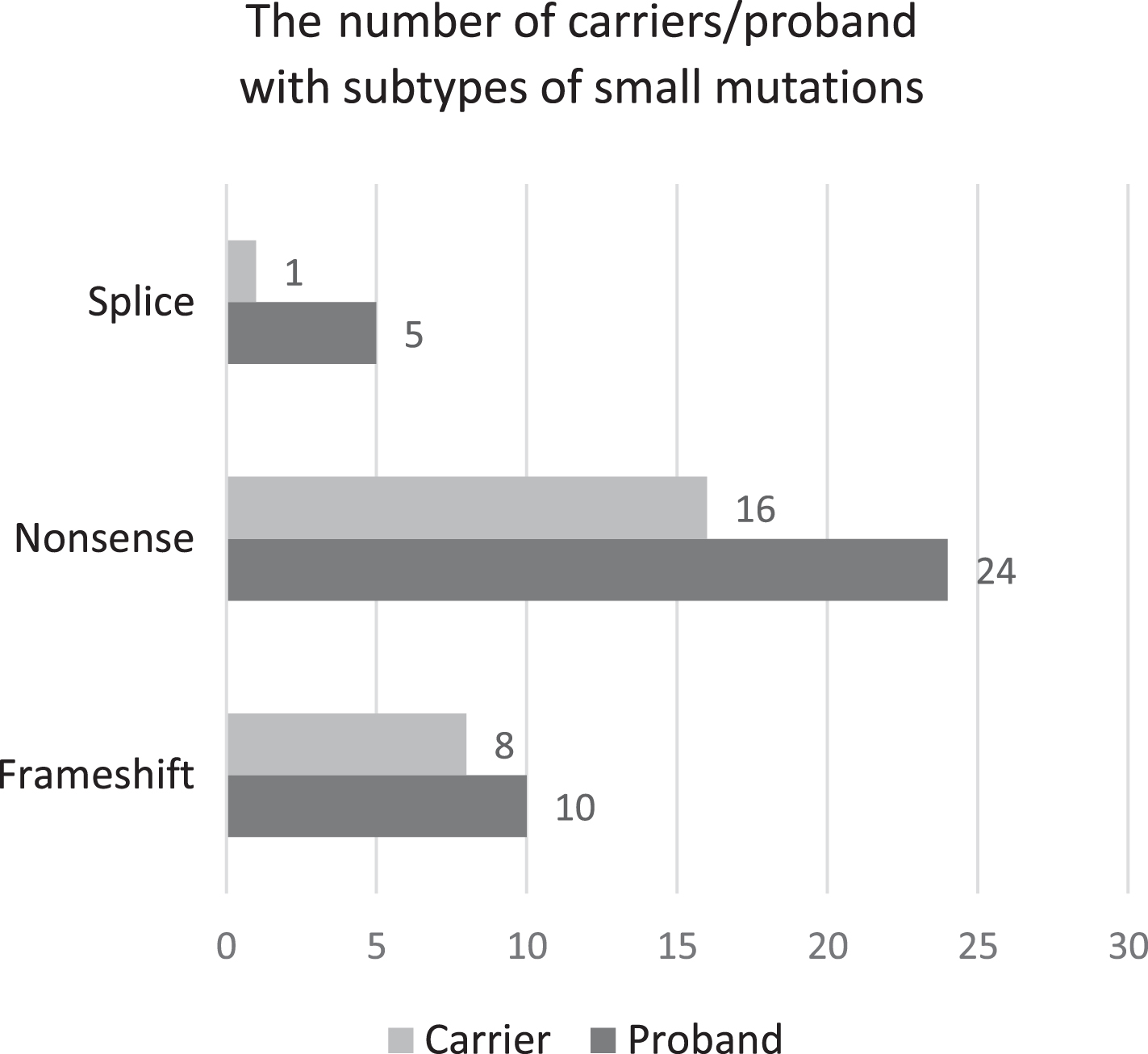
Figure representing the distribution of the various small pathogenic variants among the probands and carriers. The carrier frequency for frameshift variants was 80%, 66.6% for nonsense variants and 20% for splice site variants (P-value = 0.054).
De novo variant rate
The overall de novo variant rate is 41.8% and the rate for large deletions (52.2%) was significantly higher than other pathogenic variants.The rate for small pathogenic variants is 35.9% (Table 2).
De novo pathogenic variant rate as per the type of variant
DISCUSSION
The current report is the first study from India to address the inheritance rate of all types of pathogenic variants in DMD in a large cohort. Establishing the carrier status in mothers of DMD probands is imperative for genetic counseling, advice for future pregnancies, and identifying other female relatives who may have a risk of bearing children with DMD. The determination of carrier frequency for different pathogenic variants is needed as there is a difference in the inheritance rate among the various pathogenic variants responsible for DMD [15, 16]. This again plays a vital role while counseling. MLPA is the most reliable test to identify large deletions or duplications and is the recommended test for carriers. At the same time, the diagnosis of small pathogenic variants requires Sanger sequencing or NGS. Our study incorporated MLPA and Sanger sequencing for determining the carrier frequency based on the type of pathogenic variant in the DMD gene apart from screening for symptomatic carriers. Though there have been a few studies on DMD carriers in India, the majority of them have focused on large deletions or duplications and did not test for small pathogenic variants in significant numbers [17, 18].
In our study, we observed an overall carrier frequency of 58.2%, which is slightly lower than the expected rate of inheritance of 67% given by the formula by Haldane, who proposed that approximately one-third of patients of an X-linked recessive disorder have de novo variants and two-thirds inherit the variant from carriers [19]. It is similar to the rate reported in studies conducted in other countries [15, 21] but significantly lower than the inheritance rate of 74.9% reported by the French UMD-DMD database [22]. The rate mentioned in this database may be higher as it includes data collected from mothers of both DMD and BMD probands. Lee et al. have reported a higher overall carrier frequency in mothers of BMD probands than DMD, as BMD probands can have descendants [15].
On the other hand, a low inheritance rate of 45.8% was noted in an Australian cohort where the pathogenic variants were detected using quantitative PCR and DNA sequencing [23]. Similar inheritance rates in the range of 40– 50% have been reported in Chinese and Turkish studies by Zhong et al., and Toksoy et al., respectively, and they employed NGS and MLPA to identify carriers [24, 25]. Prior literature from India points to a further lower positive carrier rate of 40% [12, 18]. This probably is because of the smaller sample size, different techniques with varying sensitivity, the inclusion of all potential female relatives, and evaluation for large deletions and duplications only. While the positive DMD carrier frequency in the present study is 58.2%, there were 4 such families where carrier testing in mothers was negative inspite of positive X-linked recessive family history (Supplementary Table). In F35, 37 and F79 maternal uncles had history of DMD phenotype with early deaths. While we did not have the genetic data from affected maternal uncles, germline mosaicism can probably explain the lack of DMD mutation in maternal DNA extracted from peripheral blood [26]. Further, in F40 family affected younger brother had deletion of different exon 7 while proband was identified to have deletion of exons 10 and 11. This indicates a rare possibility of two de-novo deletions occurring in same family as mother tested negative for DMD deletions.
In the present study, we found that the carrier frequency for large deletions (47.8%) was significantly lower compared to the other pathogenic variants (P-value = 0.028). This is in accordance with earlier reports from Japan and Poland which have reported a similar low inheritance rate for large deletions [15, 16]. A comparison of the carrier frequency of large deletions with other pathogenic variants revealed a similar difference across several international studies (Table 3). The lower inheritance rate for large deletions is because of the difference in the origin of the large deletions and duplications compared to the small pathogenic variants [26]. Large deletions occur de novo during oogenesis due to unequal crossing over. Small pathogenic variants arise due to DNA replication or repair errors during cell division, which occur more commonly during spermatogenesis, and these paternally derived variants are transmitted through a maternal carrier, hence accounting for high carrier frequency for small pathogenic variants than large deletions [26]. In our study, a higher carrier frequency of 64.1% was observed in families with small mutations,which is similar to other studies (Table 3) [15, 25]. Among the small pathogenic variants, though substitutions resulting in nonsense mutations were more frequent among the probands, the carrier frequency for frameshift causing small insertions/deletions (80%) was much higher than either nonsense (66.6%) or splice region variants (20%).
Comparison with similar studies in other countries and India
We acknowledge that the percentage of DMD families with small mutations (39/91– 42.8%) included in this study is relatively higher in comparison to the mutation pattern reported by Polavarapu et al., in 606 Indian DMD patients [5]. This can be attributed to the fact that this study included DMD families diagnosed from 2015 when NGS testing was started as a routine diagnostic method for identifying small mutations in MLPA negative DMD patients. This led to increased identification of patients with small mutations during the last few years. Furthermore, due to the higher prevalence of positive family history in those with DMD small mutations, there appears to be an increased voluntary acceptance for carrier testing in these families.
Though the inheritance of large duplications may be similar to that of large deletions, most of the studies so far have noted a higher carrier frequency for duplications in comparison to deletions ranging from 50– 80%. Interestingly the current study detected a higher carrier frequency for large duplications (100%), with all the large duplications in the DMD probands being inherited from the mother. As we had very few probands with duplications, studies on larger cohorts will be needed for more accurate estimation.
As per literature, around 2.5– 7.8% of the DMD carriers can develop progressive muscle weakness [27]. Further, skeletal muscle damage may be seen in 2.5– 19% of DMD carriers [28]. Dilated cardiomyopathy has been observed in 7.3– 16.7% of carriers in large worldwide cohort studies [28]. It is also important to note that there is an increased frequency of cardiomyopathy with increasing age, thus highlighting the importance of follow-up in this cohort. Among the mothers in our study, we had two symptomatic carriers with progressive weakness and one with dilated cardiomyopathy. However, we could not conduct serum CK, and formal cardiac evaluation for all the carriers. These are the limitations of this study.
CONCLUSION
To summarize the findings above, 58.2% of the pathogenic variants in DMD probands were inherited. Large deletions had a significantly higher de novo variant rate of 52.2%. The carrier frequency for small pathogenic variants (64.1%) was significantly higher than that of large deletions (47.8%). Testing the carrier status of mothers has important genetic implications and is invaluable for counseling and family planning. Screening the mothers for muscle weakness and cardiomyopathy during initial visit and follow-up is recommended for appropriate early management.
Footnotes
ACKNOWLEDGMENTS
The authors would like to thank patients and their families who volunteer to participate in clinical research and give permission to use their data for analysis.
AUTHOR CONTRIBUTIONS
Conceptualization: A Nalini, Kiran Polavarapu, Divya Nagabushana
Data curation: Divya Nagabushana, Kiran Polavarapu
Formal analysis: Seena Vengalil, Saraswati Nashi, Tanushree Chawla, Veeramani Preethish-Kumar
Investigation: Kiran Polavarapu, Swetha Gunasekaran, Gautham Arunachal
Methodology: Divya Nagabushana, Priya Treesa Thomas, Arun Sadasivan, Manjusha Warrier, Muddasu Keerthipriya, Sanita Raju
Software: Kiran Polavarapu, Divya Nagabushana, Mainak Bardhan
Validation: Kiran Polavarapu, Swetha Gunasekaran, Gautham Arunachal, A Nalini, Ram Murthy Anjanappa, Mainak Bardhan
Writing review& editing: all authors.
FUNDING
The authors have no source of funding to declare.
CONFLICT OF INTEREST
The authors have no potential conflict of interest to disclose.