
Abstract
Research and drug development concerning rare diseases are at the cutting edge of scientific technology. To date, over 7,000 rare diseases have been identified. Despite their individual rarity, 1 in 10 individuals worldwide is affected by a rare condition. For the majority of these diseases, there is no treatment, much less cure; therefore, there is an urgent need for new therapies to extend and improve quality of life for persons who suffer from them. Here we focus specifically on rare neuromuscular diseases. Currently, genetic medicines using short antisense oligonucleotides (ASO) or small interfering ribonucleic acids that target RNA transcripts are achieving spectacular success in treating these diseases. For Duchenne muscular dystrophy (DMD), the state-of-the-art is an exon skipping therapy using an antisense oligonucleotide, which is prototypical of advanced precision medicines. Very recently, golodirsen and viltolarsen, for treatment of DMD patients amenable to skipping exon 53, have been approved by regulatory agencies in the USA and Japan, respectively. Here, we review scientific and clinical progress in developing new oligonucleotide therapeutics for selected rare neuromuscular diseases, discussing their efficacy and limitations.
Keywords
ABBREVIATIONS
Antisense oligonucleotide RNA-mediated interference Food and Drug Administration Induced pluripotent stem cells Urine-derived stem cells Duchenne muscular dystrophy Myotonic dystrophy type 1 Hereditary transthyretin amyloidosis Amyotrophic lateral sclerosis Spinocerebellar ataxia Spinal muscular atrophy Survival motor neuron Familial amyloid polyneuropathy Frontotemporal dementia Huntington disease Locked nucleic acid Methoxyethyl O methyl Phosphorodiamidate morpholino Peptide nucleic acid Phosphodiester Phosphorothioate Splice-switching oligonucleotide Exonic splicing enhancer Drug delivery system
SIGNIFICANCE OF OLIGONUCLEOTIDE THERAPEUTICS FOR RARE NEUROMUSCULAR DISEASES
Rare diseases are defined as those that affect fewer than 7, 4, or 5 in 10,000 people in USA, Japan, or EU, respectively. Individually, these diseases are rare; however, collectively they affect an estimated 475 million people, or approximately 6% of the global population [1]. Since orphan drug acts were passed by the USA (1983), Japan (1993) and the EU (2000), to induce pharmaceutical companies to develop therapies for rare diseases, investment and innovation for rare disease therapies have been accelerated. The rare disease market continues to expand rapidly, and current forecasts predict that worldwide orphan drug sales could increase at an annual growth rate of 17% through the year 2024 [1]. Among the 7,000 described rare diseases [2], 80% have genetic causes, and approximately 95% remain without therapeutic options [3]. For example, collectively, rare neuromuscular diseases are relatively common, with an estimated prevalence of 1,344/100,000 adults, including those identified through physician billing in Canada [4].
THERAPEUTIC OLIGONUCLEOTIDE PLATFORM
Antisense oligonucleotides (ASOs) are synthetic, single-stranded oligomers of diverse chemistries, short DNA-like molecules (15–30 nucleotides). ASOs have the potential to hybridise RNA sequence motifs to modulate gene expression levels via various mechanisms. They can be classified into two major categories: RNase H1-dependent and steric-blocking oligomers.
Development of oligonucleotide therapeutics began in the late 1970s when RNase H1-dependent oligomers were used to inhibit replication of an RNA virus [5]. To date, RNase H1-dependent ASOs, so-called gapmer ASOs, have been widely used as a means of downregulating disease-causing or disease-modifying genes. The endogenous RNase H1 enzyme recognises RNA–DNA heteroduplex substrates that are formed when DNA-based oligonucleotides bind to their cognate mRNA transcripts and catalyse degradation of immature pre-mRNAs (Fig. 1A). Cleavage at the site of ASO binding destroys the target pre-RNA, thereby silencing target gene expression. There were many obstacles to proving that the approach could be effective, and decades passed before the world’s first ASO drug, fomivirsen, was approved in 1998 to treat cytomegalovirus retinitis [6]. To date, two more RNase H1-dependent ASOs have received regulatory approval, mipomersen and inotersen.
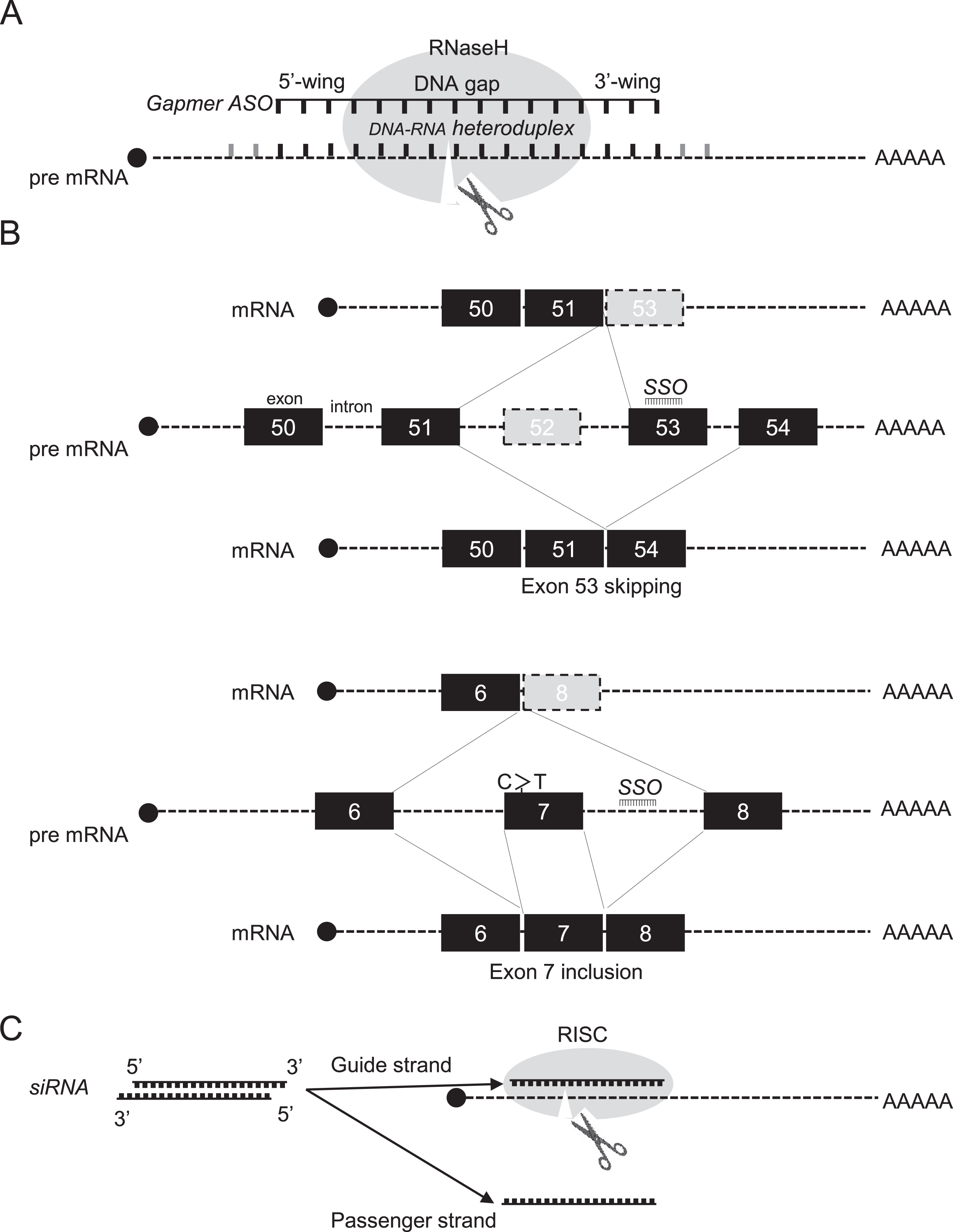
Therapeutic oligonucleotides-mechanisms of action. A. Mechanism of action for RNase H1-dependent antisense oligonucleotides (ASOs), so-called gapmer ASOs, are chimeric ASOs that contains a central block of Gapmerdesign consists of a 5′-wing followed by a central gap of 8 to 12 deoxynucleotide (DNA) monomers followed by a 3′-wing. It is an RNA-DNA-RNA-like configuration (e.g. 3-10-3). DNA and RNA strands of an DNA-RNA heteroduplex are cleaved by ribonuclease H (RNase H). premature mRNA: pre mRNA. B. Exon 53-skipping by an appropriate splice switching oligonucleotides (SSO), as indicated by an black line, restores dystrophin translational reading frame in Duchenne muscular dystrophy, who lacks exon 52 in the DMD gene leading to out-of-frame products. C to T point mutation at exon 7 of SMN2 gene induces exon 7 skipping of SMN2 in spinal muscular atrophy. Targeting the intronsplice silencer site within intron 7, by SSO mediated splice switching, induces exon 7 inclusion. C. Mechanism of siRNA-induced gene silencing. RISC: RNA-induced silencing complex. siRNA consists of a duplex of two 21-nucleotide RNAs with 19 complementary bases. The antisense strand of the siRNA (the guide strand) is complementary to a target transcript, whereas the sense strand is designated the passenger strand.
On the other hand, the most widely used application of steric-blocking or splice-switching ASOs is in modulation of alternative splicing to induce exon skipping and exon inclusion, that is, selectively exclude or retain specific exons, respectively. In these cases, exon skipping uses steric block splice-switching ASOs that hybridise to pre-RNA sequence motifs and prevent assembly of the spliceosome, leading to restoration of translatable mRNA transcripts (Fig. 1B) [4]. Splice-switching ASOs are commonly designed to bind to 5′ or 3′ splice junctions and to sterically block access of splicing factors to their target sites, altering pre-mRNA splicing (Fig. 1B) [14, 23]. ASOs may also bind to exonic/intronic splicing enhancers or silencers to either promote or block splicing. As a result, spliceosomes are diverted to other sites, and the reading frame is restored, leading to production of alternative mRNAs and restoration of protein expression. To date, the exon skipping approach using splice-switching ASOs has seen wider application to several rare neuromuscular diseases [7], including DMD [8], merosin-deficient congenital muscular dystrophy [9], limb-girdle muscular dystrophy [10], Pompe disease [11], cardiomyopathies [12], cystic fibrosis [13] and laminopathies [14].
Finally, another approach called RNA-mediated interference (RNAi) to target RNA was developed using the nematode, Caenorhabditis elegans [15], leading to RNAi-mediated gene therapy using double-stranded small interfering RNAs (siRNAs). siRNA consists of a duplex of two 21-nucleotide RNAs with 19 complementary bases and terminal 2-nucleotide 3’ overhangs) [16]. The antisense strand of the siRNA (the guide strand) is complementary to a target transcript, whereas the sense strand is designated the passenger strand. siRNAs guide Argonaute 2 protein (AGO2), a component of the RNA-induced silencing complex (RISC), to complementary target transcripts. Complete complementarity between the guide siRNA and the target transcript results in cleavage of the target, opposite positions 10–11 of the guide strand and catalysed by AGO2, leading to gene silencing (Fig. 1C). Numerous variations of the archetypal siRNA design have provided benefits in reducing passenger strand activity and/or improved potency. These include Dicer substrate siRNAs, small internally segmented siRNAs, self-delivering siRNAs (asymmetric and hydrophobic), and single-stranded siRNAs [17–22].
PRACTICAL APPLICATIONS OF OLIGONUCLEOTIDE THERAPEUTICS FOR RARE NEUROMUSCULAR DISEASES
Precision genetic therapeutics using oligonucleotides, such as synthetic ASOs and siRNAs are emerging as a new pharmacological modality for rare neuromuscular diseases. Recent advances in genomics, oligonucleotide synthesis, delivery and manufacturing, and increased collaborations and licensing opportunities have all contributed to a surge in development of synthetic oligonucleotide drugs. ASOs, in particular, are becoming more widespread as gene therapeutics, and chemical modifications that enhance their properties make them attractive candidates for drug development [23]. Surprisingly, more than 150 such oligonucleotide drugs are in various stages of development [22]. Notably, life spans of infants suffering from spinal muscular atrophy (SMA) types 1, 2 and 3 have been prolonged due to treatment with nusinersen, compared with untreated children. There have also been improvements in their gross motor development after ASO-based treatment, as measured using the Hammersmith Infant Neurological Examination [24]. Moreover, oligonucleotide drugs are in development for rare diseases such as myotonic dystrophy type 1 (DM1), amyotrophic lateral sclerosis (ALS), and polyglutamine (PolyQ)-related diseases, including spinocerebellar ataxia (SCA) and Huntington’s disease (HD), afflictions for which previously there was no hope of treatment [25]. To date, nine ASOs and two siRNAs including eteplirsen [26], golodirsen [27] and viltolarsen [28] for treatment of Duchenne muscular dystrophy (DMD), inotersen and patisiran for hereditary transthyretin amyloidosis (ATTR) [29] and givosiran for acute hepatic porphyria [30] have received U.S. Food and Drug Administration (FDA) approval.
MODELS FOR HUMAN RARE NEUROMUSCULAR DISEASES CONTRIBUTE SIGNIFICANTLY TO MODERN MEDICINE
Preclinical development is essential to transition oligonucleotide drug discovery in the laboratory to initiation of human trials. The results of preclinical pharmacology and toxicology testing in animal and cellular models leads to oligonucleotide drug candidate selection for rare diseases. Modelling diseases in experimental animals via genetic engineering is indispensable in studying pathogenic mechanisms and developing therapies for rare neuromuscular diseases. Those include DMD [31–35], DM1 [36], ALS [37], and SCA [38]. However, animal models, including rodents, canines, and primates, have limitations in translational research in evaluating therapeutic efficacy, metabolic profiles, and safety profiles of drug candidates, because of differences in nucleic acid sequences, metabolism, and physiological functions between humans and other animals. These limitations have resulted in failures to predict clinical efficacy in human patients from experimental findings in animal models of rare diseases, reducing the value of preclinical animal models. To overcome such limitations, genetically humanised animal models that allow assessment of ASO drugs, such as hDMD/mdx, hDMDdel52/mdx, hDMDdel45 D2/mdx and SMND7, could facilitate development of ASOs that target human disease-relevant transcripts for rare neuromuscular diseases [39, 40]. On the other hand, it is not pragmatic to develop many different models to perform pre-clinical studies for each and every ASO, considering that oligonucleotide therapeutics are mutation-dependent.
In contrast, clinically relevant cellular models of human diseases recapitulating phenotypes specific to those diseases, are fundamentally important. The emergence of direct or indirect, human somatic cell reprogramming technologies may overcome this limitation, because they enable use of disease- and patient-specific cellular models as enhanced platforms for drug discovery and autologous cell-based therapy. Induced pluripotent stem cells (iPSCs) and urine-derived stem cells (USCs) are increasingly employed to recapitulate pathophysiology of various human diseases [41]. Recent cell-based modelling approaches utilise highly complex differentiation systems, including three-dimensional systems that faithfully mimic human tissue- and organ-level dysfunctions and that could be used for oligonucleotide drug development [42].
OLIGONUCLEOTIDE CHEMISTRY FOR TREATMENT OF RARE DISEASES
Initially, unmodified ASOs were subject to degradation by endonucleases and exonucleases when introduced into biological systems, limiting their therapeutic potential. Modifications to the phosphodiester backbones of ASOs yielded analogues such as phosphorothioate, methylphosphonate, and phosphoramidate, which belong to the first generation of ASOs (Fig. 2A) [43, 44]. Modifications to ASO deoxyribose yielded compounds including 2’-O-methyl (2’-OMe), 2’-O-methoxyethyl (2’-MOE) and 2’-fluoro that comprise second-generation ASOs (Fig. 2B) [43, 44]. Third generation ASO modifications include phosphorodiamidate morpholinos (PMOs), peptide nucleic acids (PNAs), and tricyclo-DNAs [43, 44] as well as ethylene-bridged nucleic acids (ENAs) and locked nucleic acids (LNAs), a class of nucleic acid analogues with a methylene bridge connecting the ribose 2’-oxygen and 4’-carbon [45] (Fig. 2C). Additionally, the non-bridging phosphate oxygen replacement with a sulphur atom in the phosphodiester linkage generates a phosphorothioate backbone (PS), which enhances binding to plasma proteins and resistance to nuclease activity, prolonging the half-life of the ASO [46]. PS ASOs typically include the two stereoisomers, including Rp and Sp, which differ in their binding affinities and susceptibility to degradation. Stereo-pure ASOs from WAVE Life Sciences have mitigated toxicity while preserving function [47].
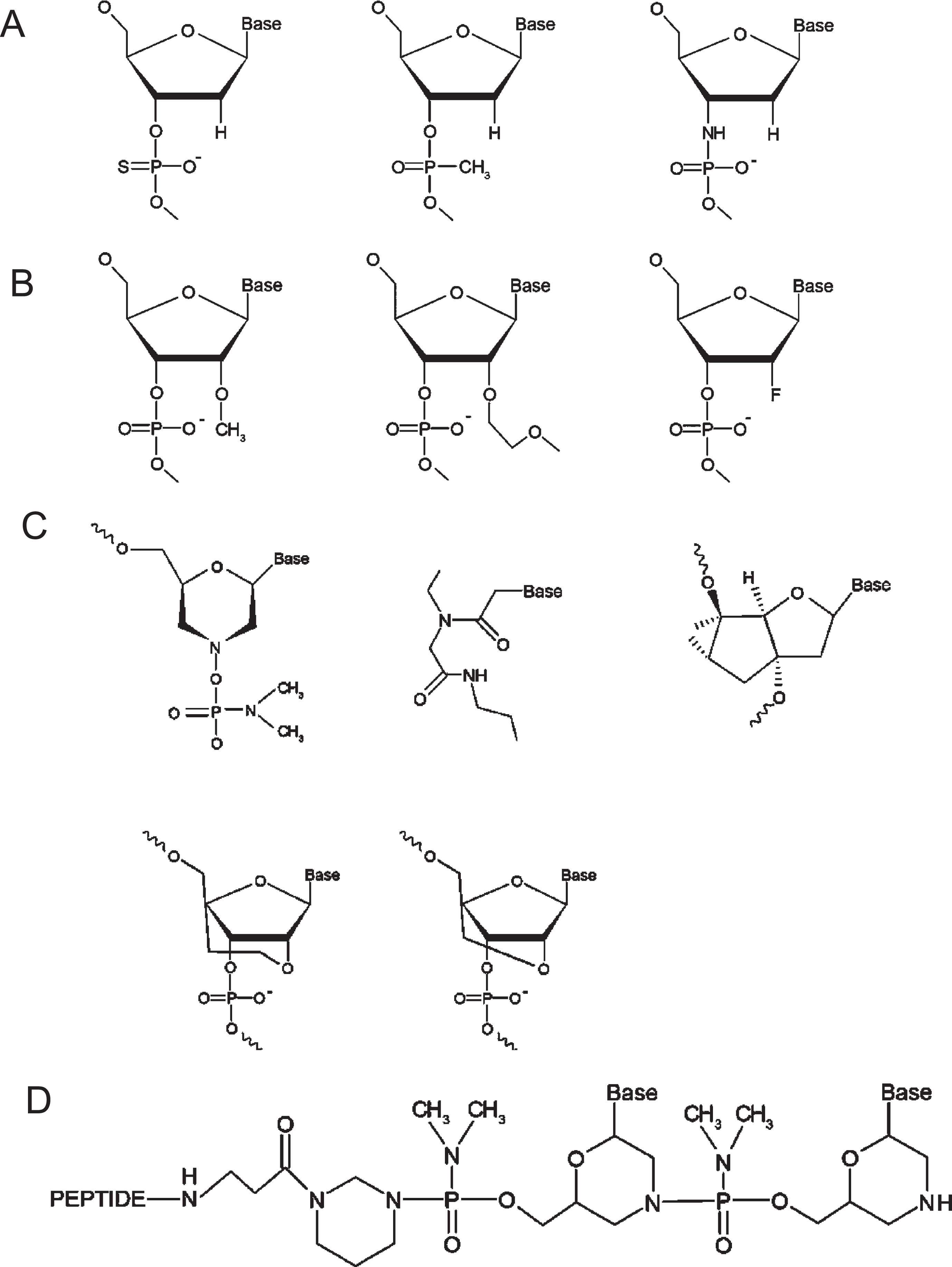
Chemical structures of therapeutic oligonucleotides. A. First generation ASOs, including phosphothioate, methylphosphonate, and phosphoramidate (from left to right). B. Second generation ASOs, including 2′-O-methyl (2′-OMe), 2′-O-methoxyethyl (2′-MOE) and 2′-fluoro (from left to right). C. Third generation ASOs, including phosphorodiamidate morpholinos (PMO), peptide nucleic acids (PNA), and tricyclo-DNAs (upper, from left to right), as well as ethylene-bridged nucleic acids (ENA) and locked nucleic acids (LNA)(lower, from left to right). D. The new generation of peptide-conjugated PMOs (PPMOs). Figure 2 adapted from Tsoumpra MK, et al. EBioMedicine. 2019;45:630–45.
PMOs are among the most promising ASOs, in which the deoxyribose ring is replaced with a morpholine ring, albeit the charged phosphodiester inter-subunit linkage is replaced with a non-ionic phosphorodiamidate linkage. This non-ribose-based modification renders PMOs immune to nuclease activity, but their non-ionic nature minimises nuclear uptake. Conjugation of a peptide to the PMO backbone generates peptide-conjugated PMOs (PPMOs) that exhibit dramatically improved pharmacokinetic profiles, while minimising toxicity, especially nephrotoxicity (Fig. 1D) [48, 49]. When tested in animal models of DMD, PPMOs demonstrate efficacious exon skipping in target skeletal and cardiac muscles and prolonged duration of dystrophin restoration after a treatment regime [50].
Another approach uses synthetic, noncoding RNA molecules called siRNA, with the potential to silence gene expression. siRNAs usually comprise 21-bp duplexes composed of passenger/guide siRNAs, which are sufficient to form stable guide siRNA-target mRNA duplexes, provided that the guide siRNA is recognised by RISC (Fig. 1E) [51, 52]. siRNA-based drugs require chemical modification or formulation of the passenger siRNA to promote stability, minimise innate immunity, and enable delivery to target tissues, including muscles and central nervous system. The great potential of siRNA-based drugs for rare neuromuscular diseases is exemplified by patisiran for ATTR treatment, which has been approved in the USA and the EU [53, 54].
OLIGONUCLEOTIDE THERAPEUTICS FOR DMD
Duchenne muscular dystrophy (DMD), the most common and severe form of muscular dystrophy, is caused by a mutation in the DMD gene [55]. It exhibits X-linked inheritance and occurs in 1 of 4,000-5,000 male births worldwide [56]. In 2/3 of all cases, the mother carries the DMD gene abnormality, and in the remaining 1/3, the DMD gene in the mother’s oocyte is newly mutated. Female carriers are most often asymptomatic, but in rare cases, they have elevated serum creatine kinase (CK) levels or slight muscle weakness [57].
Considerable effort has been devoted to developing genetic therapies to restore dystrophin expression in muscles of DMD patients. The leading therapeutic approach is exon skipping, whereby splicing of the dystrophin pre-mRNA transcript is modulated to restore the translation reading frame, rescuing dystrophin expression. The concept of exon-skipping due to an intraexon deletion in the DMD gene was first demonstrated in DMD skeletal muscle cells in 1991 [58]. The exon-skipping proof-of-concept has been demonstrated in dystrophin-deficient myoblasts [59–61], dystrophin-deficient mice [62, 63] and muscular dystrophy dogs [33, 35], and has subsequently become established as an efficacious assay system for exon-skipping treatment using dermal fibroblasts [64] or urine-derived cells from DMD patients [65, 66]. PMOs are being researched as potential therapeutic agents for DMD. In 2016, eteplirsen, a PMO drug, was approved by the FDA to treat DMD [26]. Eteplirsen theoretically treats up to 13% of DMD patients by skipping exon 51 and restoring the reading frame of DMD transcripts. Because other mutations could also cause DMD, PMOs specific to the various offending reading frames are under constant development. Golodirsen (Sarepta Therapeutics) and viltolarsen (Nippon Shinyaku and NS Pharma) are PMOs that target exon 53 on the DMD primary transcript. Theoretically, 7.7% of DMD patients would benefit from golodirsen or viltolarsen treatment [27, 68]. Provided that eteplirsen is approved, multiexon skipping with PMOs targeting exons 51 and 53 could restore the reading frame of mRNA, which could be applied to an additional 8.1% of DMD patients.
In phase 1/2 clinical trials (NCT02310906) of golodirsen, 25 boys (ages 6–15) with DMD amenable to exon 53 skipping received 30 mg/kg golodirsen and a significant increase in exon 53 skipping was associated with dystrophin protein expression at week 48, with a mean percent normal dystrophin protein standard of 1.019% (range, 0.09%–4.30%) [69]. In the dose-finding phase 2 randomised clinical trial of viltolarsen (NCT02740972), 16 boys (ages 4–9) with ambulant DMD amenable to exon 53 skipping received 40 mg/kg or 80 mg/kg of viltolarsen once a week for 24 weeks, and 14 boys reached dystrophin levels higher than 5.3-5.4% of normal [70]. Very recently, golodirsen was approved in the USA and viltolarsen in Japan and the USA. It should be noted that eteplirsen, golodirsen, and viltolarsen were all approved based only on dystrophin levels, not on functional effects on muscles. Although no severe adverse effect has been observed in the clinical development program, monitoring of renal function is prudent, given the findings of nephrotoxicity seen with other ASOs [48, 71–73].
OLIGONUCLEOTIDE THERAPEUTICS FOR SMA
Spinal muscular atrophy (SMA), one of the most common genetic causes of infantile death, is caused by a mutation in the survival motor neuron (SMN1) gene, with an incidence of 1:10,000 [74]. Subsequent reduction in expression of the widely expressed SMN results in selective loss of spinal cord motor neurons. There are primarily 4 types of SMA (type 1-4), classified accordingly to a person’s age at onset and the disease’s clinical course. Type I SMA patients have 2 copies, type II patients have 3 copies, type III have 3 or 4 copies, and type IV have 4 copies of the SMN2 gene [75]. The number of SMN2 copies is inversely correlated with SMA disease severity.
SMA type 1, the most frequent and severe form, results in lethal infantile paralysis, but all forms of SMA cause severe disability. The SMN2 gene, which is variably spliced and has a similar structure to SMN1, except for excluding exon 7 in most transcripts, produces small amounts of full-length SMN protein, below the threshold required for spinal motor neurons, but sufficient for normal function in most cells. After the discovery of the intronic splicing silencer N1 (ISS-N1), ISS-N1-targeting ASOs to stimulate SMN2 exon 7 inclusion have been developed to include exon 7 in the mature SMN2 transcript, thereby increasing production of SMN protein.
Nusinersen, a modified 2′-MOE, required intrathecal injections and was the first ASO approved by the FDA in 2016. For infants with SMA who had symptom onset after 6 months of age, 57% of infants in the nusinersen cohort, compared with 26% in the control cohort, had an increase in the Hammersmith Functional Motor Scale Expand score of at least 3 points (p < 0.001) from baseline to month 15, giving encouraging evidence of efficacy [76] (NCT02292537). Though slightly off-topic, in 2019, Zolgensma, an adeno-associated virus 9 vector that carries a normal copy of the SMN1 cDNA to its target motor neurons, was approved by the FDA to treat all SMA types in newborns and toddlers up to age 2 through a single intravenous administration (NCT03505099). Risdiplam, an orally administrated SMN2 splicing modifier to increase levels of functional SMN protein for all types of SMA [77] (Roche and Genentech), demonstrated significant motor milestone improvement in SMA type 1 (NCT02913482), as well as SMA types 2 and 3 (NCT02908685). It has been approved by the U.S.the FDA, European Commission and Japan as the first and only at-home treatment for SMA.
OLIGONUCLEOTIDE THERAPEUTICS FOR DM1
Myotonic dystrophy type 1 (DM1) is one of the most common forms of muscular dystrophy in adults, with an incidence of 1:15,000. This is a multisystemic disorder characterised by progressive muscle wasting, as a major feature of DM1, and by myotonia, cardiac conduction defects, endocrine deficiencies, and cognitive impairments. It can also affect infants and results in severe disability and significantly shortened life span. No treatment is presently available. DM1 is caused by the expansion of a CTG repeat in the 3′-untranslated region of the Dystrophia Myotonica Protein Kinase (DMPK) gene. CUG-expanded DMPK mRNA, containing abnormally expanded CUG repeats, sequesters muscle blind-like (MBNL) alternative splicing regulators in ribonuclear inclusions (RNA foci) and interferes with alternative splicing of pre-mRNA that directly causes DM1. Oligonucleotides targeting pathologic RNAs show promise in DM1 treatment by targeting toxic CUG expansions and restoring the function of MBNL proteins, which mediate pre-mRNA alternative splicing regulation sequestration, contributing to DM1 [78]. This approach has been tested in a safety and tolerability study of multiple doses of ISIS-DMPKRx (Ionis Pharmaceuticals), ASOs which recruit RNAse H to cleave and degrade the pre-RNA, in adults with DM1 (NCT02312011); however, systemic administration of the selected naked ASO did not achieve sufficient concentrations in skeletal muscle of patients to have marked clinical benefits. Successful delivery of ASOs must be achieved to realise therapeutic effects for DM1 [79].
OLIGONUCLEOTIDE THERAPEUTICS FOR ALS
Amyotrophic lateral sclerosis (ALS) is an aggressive disease of motor neurons and one of the most common neurodegenerative diseases, with an incidence of 2 per 100,000 population worldwide. It is a relentlessly progressive and incurable motor neuron disease in which the average survival is 2 to 3 years from the onset and for which effective treatments are desperately needed. Approximately 12% to 15% of patients with ALS carry a disease-determining genetic mutation [37]. There seem to be many biological triggers of ALS, given that mutations in more than 20 different genes have been implicated. Superoxide dismutase 1 (SOD1) mutations, which cause a toxic gain of function associated with misfolding of mutant SOD1. The pathogenic hexanucleotide repeat expansion in the first intron of the chromosome 9 open reading frame 72 (C9ORF72) gene account for 60 to 70% of mutations that cause familial ALS and up to 10% of all ALS cases, as well as a substantial fraction of cases of the neurodegenerative disorder, frontotemporal dementia [80]. Their discoveries as causative agents of ALS facilitated the development of new therapies directed at these mutations.
ASOs delivered intrathecally enter the central nervous system, including motor neurons, and block production of both normal and misfolded mutant SOD1 protein, which were tolerated in rodent models [37]. Rodent ALS models treated intrathecally with tofersen, 2′-MOE gapmer ASO, exhibited reduced levels of mutant SOD1 protein and extended survival [37]. Following promising phase 1 results with tofersen, which showed safety, tolerability, and significantly reduced SOD1 in cerebrospinal fluid of 50 participants, phase 3 trials of tofersen (Biogen) in SOD1 ALS patients are ongoing (clinicaltrials.gov: NCT02623699).
C9ORF72 is likely to have diverse cellular functions, making targeted knockdown of mutant transcripts a promising clinical treatment, although the precise mechanism has yet to be determined. A phase 1/2 study to evaluate safety and toxicity of the ASO, IONIS-C9Rx, which is an RNAse H oligomer that selectively inhibits mutant C9ORF72 transcripts, is presently underway in ALS patients (clinicaltrials.gov: NCT03626012) [37]. Recently, IONIS-C9Rx was granted a fast-track designation in the USA, which will facilitate expedited development and review of the drug.
OLIGONUCLEOTIDE THERAPEUTICS FOR HUNTINGTON’S DISEASE
Huntington’s disease (HD) is a dominant, inherited rare neurodegenerative disorder caused by a CAG trinucleotide expansion in the HTT gene, responsible for the pathologic mutant huntingtin protein, with an incidence of 0.38 per 100,000 [81]. Tominersen (Roche) (previously IONIS-HTTRx and RG6042), a 2′-MOE gapmer targeting the HTT transcript, binds to HTT mRNA, resulting in endogenous RNase H-mediated degradation of HTT mRNA, thus inhibiting translation of huntingtin protein. Preliminary results from a phase 1/2 study of tominersen (NCT02519036), showing reduced mutant huntingtin in cerebrospinal fluid, are promising [82]. Unfortunately, however, Roche has discontinued dosing in the phase 3 trial of its investigational therapy tominersen in adults with HD (NCT03761849), although a phase I pharmacokinetic/pharmacodynamic study of tominersen (NCT04000594) and the observational HD natural history study (NCT03664804) will continue. In parallel, Wave Life Sciences had been conducting the PRECISION-HD1 and -HD2 phase 1b/2a clinical trials of stereopures ASOs, WVE-120101 and WVE-120102, designed to selectively knockdown the mutant HTT mRNA transcript that contains the single nucleotide polymorphism (SNP) rs362307 in exon 67 and the SNP rs362331 in exon 50 of the HTT transcript (NCT03225833). The company has recently stopped developing the two ASOs due to no significant change in mutant huntingtin protein in adults with HD, but it will start a new clinical trial of new ASO WVE-003 for HD, which has novel chemical modifications.
OLIGONUCLEOTIDE THERAPEUTICS FOR SPINOCEREBELLAR ATAXIA
Various neurodegenerative diseases, including polyglutamine diseases, share a common pathological mechanism involving abnormal accumulation of misfolded proteins in the central nervous system, and exon skipping is also used as a potential therapy for several SCAs. Among those, SCA3, the so-called Machado-Joseph disease, is caused by a CAG repeat expansion, encoding a polyQ repeat, in exon 10 of the ATXN3 gene, with an incidence of 3:100,000 [83]. Different approaches have been used to develop SCA3 ASOs. These have included allele-specific ASOs to the expanded CAG repeat that sterically block translation, splice-switching 2′-MOE ASOs that exclude exon 10, encoding the expanded repeat, and 2′-MOE gapmer ASOs that target wild-type and mutant ATXN3 alleles [84]. A transgenic monkey model of SCA3, recapitulating progressive neurological symptoms, may accelerate oligonucleotide drug development for the disease [85].
INDIVIDUALISED OLIGONUCLEOTIDE THERAPEUTICS FOR ULTRA-RARE NEUROMUSCULAR DISEASES
Among rare diseases, those that affect fewer than 1 in 50,000 people are defined in the EU as ultra-rare diseases. The emerging N-of-1 trial of oligonucleotide therapeutics has excellent potential to provide evidence of effectiveness for ultra-rare neuromuscular diseases [86]. It could be an ultimate strategy for precision genetic medicine because oligonucleotide sequences are highly customisable to specific mutations, and oligonucleotide drugs have excellent safety profiles and potential for rapid, inexpensive and scalable manufacturing development. Milasen, designed to treat Mila Makovec, who suffered from neuronal ceroid lipofuscinosis 7, a fatal form of Batten disease, was the first personalised ASO approved by the FDA and examplifies the potential of ASO treatment of those with ultra-rare disorders [87]. Some other individualised ASO drugs have been created and requested regulatory approval for an N-of-1 clinical trial, including Jacifusen for rare FUS-linked familial ALS by Ionis and a drug for ataxia-telangiectasia [88]. These confirm that oligonucleotide therapeutics have far greater potential for treating ultra-rare diseases than current drugs and offer priceless hope to patients and their families.
CHALLENGES IN OLIGONUCLEOTIDE THERAPEUTICS FOR RARE NEUROMUSCULAR DISEASES
Oligonucleotide therapeutics for rare neuromuscular diseases have reached a technological level sufficiently effective for drug approval if appropriate target oligonucleotide sequences, tissues and diseases are selected. Future challenges involve the appropriate selection of target tissues and diseases. For this purpose, the human RNA database, with next-generation sequencing, which includes pre-mRNAs of full length and noncoding RNAs, has been developed. The data are routinely used to assess efficacy and safety of oligonucleotide drugs, accelerating drug development. Currently, highly precise RNA retrieval technology to extract sequences complementary to oligonucleotides has been established, which is essential to predict hybridisation-dependent off-target effects in silico to circumvent collateral oligonucleotide toxicity in vivo. Recently, we developed eSkip-Finder (https://eskip-finder.org) to help researchers identify effective splice switching ASOs, the first web-based resource using machine-learning algorithms [89].
Future challenges also involve development of therapeutic oligonucleotides coupled with delivery technologies and/or directed against specific cell types or tissues (active targeting). Oligonucleotides typically accumulate in the liver, kidney, spleen, and bone marrow, and such accumulations may cause tissue or organ damage. Notably, it is critical to achieve sufficient penetration and exposure of oligonucleotide drugs in skeletal muscle and then to improve exposure in heart and brain tissues to treat specific rare neuromuscular diseases. Currently, local intrathecal delivery of oligonucleotides to the CNS has been established, although deep brain structures are not always effectively targeted even by this route of administration. To enhance efficacy of oligonucleotide drugs, for example, ASOs may be conjugated to a new class of peptide ligand, known as cell-penetrating peptides, which offer great possibilities for oligonucleotide delivery, with very high efficiency to affected tissues [43]. Currently, preclinical and clinical development of novel ASOs conjugated to arginine-rich, cell-penetrating peptides are being developed for DMD (SRP-5051) by Sarepta Therapeutics and also for related neuromuscular diseases by PepGen [49]. In addition, antibody ASO conjugates, which could allow targeted muscle uptake, are being developed to treat life-threatening rare neuromuscular diseases (Avidity Biosciences and Dyne Therapeutics) [67]. Moreover, glucose transporter-1-based siRNA delivery strategy, which could boost glucosylated nanocarriers crossing the blood-brain barrier into the brain, might be promising [90].
Long-term treatment with oligonucleotides may provoke immune responses to newly expressed proteins. For instance, restored very high levels of dystrophin induced by exon skipping in a short period resulted in an immune response [91]. Although such potential contraindications of ASO drugs must be thoroughly investigated in patients with DMD and SMA during post-marketing surveillance, shorter dystrophin isoforms or SMN protein in SMA might induce immune tolerance in patients with DMD and SMA, respectively.
Finally, studies involving healthy volunteers to provide the scientific evidence for determining whether an investigational ASO drug is safe and effective are critical with rare diseases, because it could be challenging to find enough patients to gather meaningful data. Considering that patients and care givers are the end-users of developing oligonucleotide therapeutics for rare neuromuscular diseases, patient and public involvement (PPI) might help to improve enrollment in clinical trials [90] if patients are involved in discussions and actions about commissioning and regulatory processes. Patients and their supporters could collaborate with other stakeholders, including companies, regulators, and policymakers, to ensure access to effective medicines [92].
CONCLUSION
Although the most significant limitation of clinically used ASOs or siRNA is their poor delivery systems targeting the neuromuscular system, current oligonucleotide therapeutics have successfully treated rare neuromuscular diseases. As of May 2021, 4 types of RNase H1-dependent ASOs, 6 types of steric-blocking (splice switching) ASOs, 4 types of siRNA, and some other oligonucleotide-based drugs have been approved (Table 1). At present, there are several dozen candidates in late stages of development. Because oligonucleotide drugs can target noncoding RNAs as well as coding RNAs, the number of rare neuromuscular diseases targeted by oligonucleotide therapeutics is expected to increase steadily, leading to further clinical successes.
EMA, European Medicines Agency; JP PMDA, Japan Pharmaceuticals and Medical Devices Agency; FDA, Food and Drug Administration; PS, phosphorothioate; PO, phosphodiester; siRNA, small interfering RNA; single-stranded DNA; ssDNA, dsDNA, double-stranded DNA; 2′-F, 2′-fluoro; GalNac, N-acetylgalactosamine; IVT, intravitreal injection; SC subcutaneous, IT, intrathecal; IV, intravenous; CNS, central nervous system; LNP, lipid nanoparticle; 2′-MOE, 2′-O-methoxyethyl; 2′-OMe, 2′-O-methyl; PMO, phosphorodiamidate morpholino oligonucleotide; NA, not applicable. Oligonucleotide drugs for neuromuscular diseases are denoted in red.
CONFLICT OF INTEREST STATEMENT
YA declares no competing financial interests. MJAW is a founder and shareholder of PepGen Ltd. and Evox Therapeutics, dedicated to commercialising peptide-enhanced therapeutic oligonucleotide delivery and extracellular vesicle therapeutics, respectively.
Footnotes
ACKNOWLEDGMENTS
I thank Dr. Steven D. Aird for English editing and Dr. William Valentine for scientific advice.