
Abstract
Duchenne (DMD) and Becker muscular dystrophies (BMD) are rare neuromuscular disorders caused by mutations in the dystrophin gene and failure in its protein production. The absence or the reduced expression of dystrophin render muscles prone to damage, including the cardiac and respiratory muscles with reduced life expectancy. Careful planning for clinical trials will require a sufficient number of confirmed cases to meet the inclusion criteria. National registries for rare disorders serve as an essential tool for personalized medicines or mutation-specific trials to facilitate patient recruitment. The Iranian Registry of DMD and BMD (IRDAB) collects detailed molecular data of Iranian DMD/BMD patients and carriers according to the TREAT-NMD Global Neuromuscular Network guidelines. As of March 2020, five hundred and twenty-two cases are registered. The registry incorporates multi-level web and database technologies, where registrants can access their data and compare it to the cumulative data. The registry’s objectives are to recruit eligible patients for clinical trials and provide sufficient data for the national program of disease surveillance and social planning. Furthermore, the registry provides accurate epidemiological data, phenotype/genotype correlation, and evaluate the standards of care in Iran.
ABBREVIATIONS
Becker Muscular Dystrophy Duchenne Muscular Dystrophy Intermediate muscular dystrophy Electronic Health Record System Iranian Registry of Duchenne and Becker muscular dystrophies Multiplex Ligation-dependent Probe Amplification Polymerase Chain Reaction treat Neuromuscular Disease Global Network Non-governmental association Creatine phosphokinase Structured query language Secure sockets layer
INTRODUCTION
DMD and BMD are X-linked neuromuscular disorders caused by mutations in the dystrophin gene [1]. DMD is a lethal condition affecting approximately one in every 3,500 male births and caused by out-frame mutation disrupting the gene reading frame resulting in a non-functional protein leads to progressive skeletal, cardiac, and respiratory muscle weakness. With no treatment, boys lose ambulation in their early teens and have reduced life expectancy [2, 3]. BMD, the milder form, affects 1/17,000 male births due to an in-frame mutation resulting in partially functioning protein [4].
Registries are an essential resource for clinical trials; understanding each population’s specific characteristics is crucial for the development of new therapeutic strategies. TREAT-NMD Global Neuromuscular Network had played an essential role in developing DMD/BMD national registries through integrated international activities, developing a network of researchers, and setting up a coordination center for international clinical trials [9, 10]. Currently, more than 50 national and regional DMD/BMD registries are in connection with TREAT-NMD [11].
IRDAB was established in 2010; one of the registry’s primary purposes is to effectively help recruit Iranian DMD/BMD patients into national/international clinical trials [12]. International collaboration is essential, and establishing a central database will facilitate research by recruiting more patients and speeding the development of new therapies. Furthermore, one of our registry’s main objectives was to identify and characterize the total number of DMD/BMD patients nationwide and have epidemiological disease surveillance that allowed the development of our national health care and social planning program.
In this paper, we introduce the structure and function of the DMD/BMD registry in Iran, describing the data collection system and the preliminary analysis.
MATERIALS AND METHODS
Patients
IRDAB collects the data of genetically confirmed DMD/BMD patients and carriers; however, patients do not necessarily require a genetic test for registry inclusion. We include diagnosed patients through a lack of dystrophin in muscle biopsy after the approval of a neuromuscular specialist. Patients/caregivers enter their data after being confirmed by a neuromuscular specialist.
We excluded in this study patients with no genetic or pathological diagnosis. A flow diagram for data entry to the registry is shown in (Fig. 1).
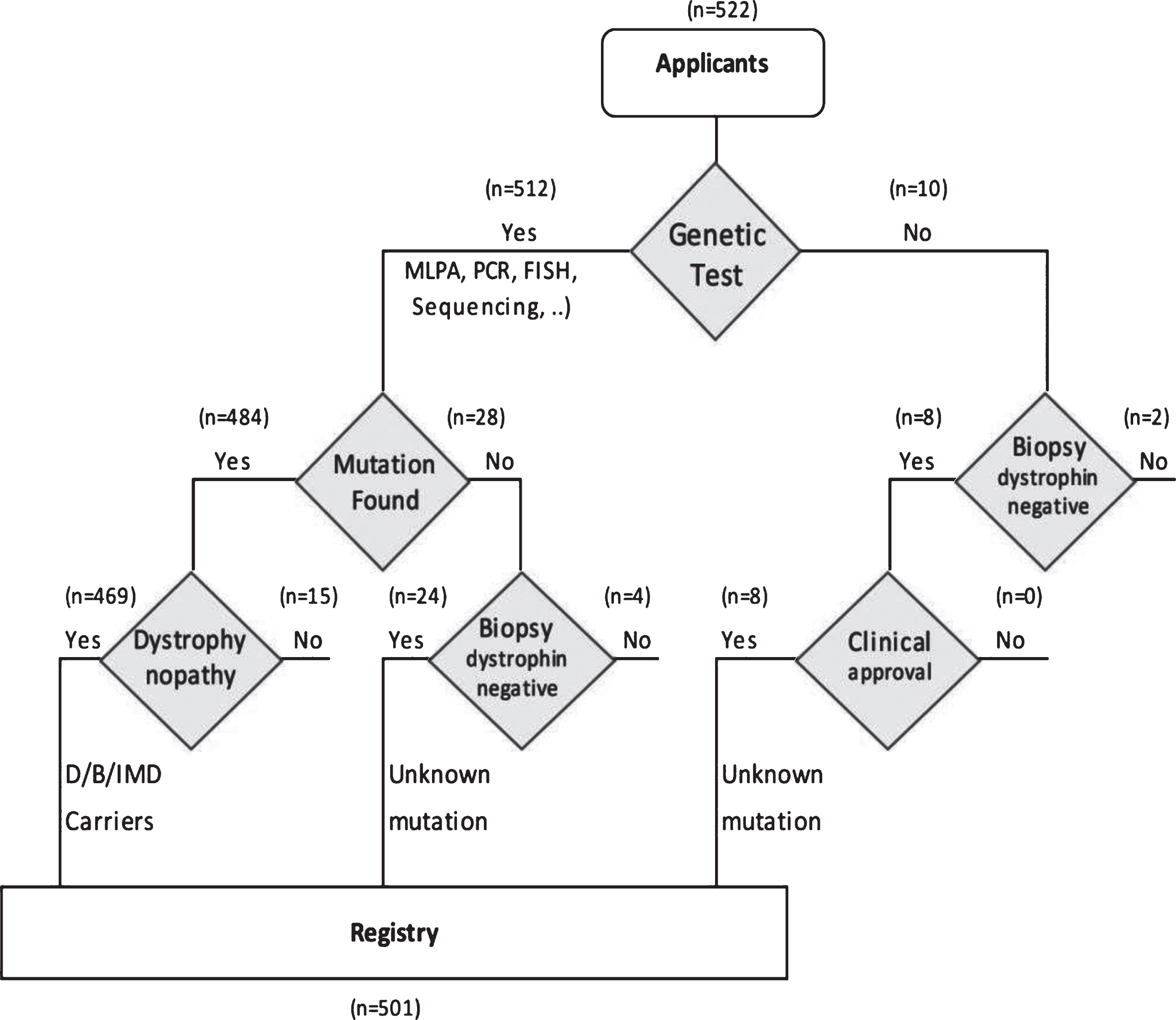
Inclusion criteria in the registry.
Establishment of registry, organization, and management
Iran Ministry of Health funds IRDAB, which is registered under Project Number 301/700, developed and managed by the lead researcher at the Pasteur Institute of Iran. The steering committee is composed of researchers, physicians, and representatives of NGOs. This registry includes information on DMD/BMD patients and carriers living in Iran. The registry includes data from nine centers in Tehran, two in Isfahan, one in Tabriz and one in Mashhad, and two NGOs. The data collection framework and methods is in adaptation with the TREAT-NMD Global Neuromuscular Network. The country and registrants’ population distribution in each province of Iran is demonstrated in (Fig. 2). Most of the registered cases are from the capital Tehran, Isfahan, and the northern cities.
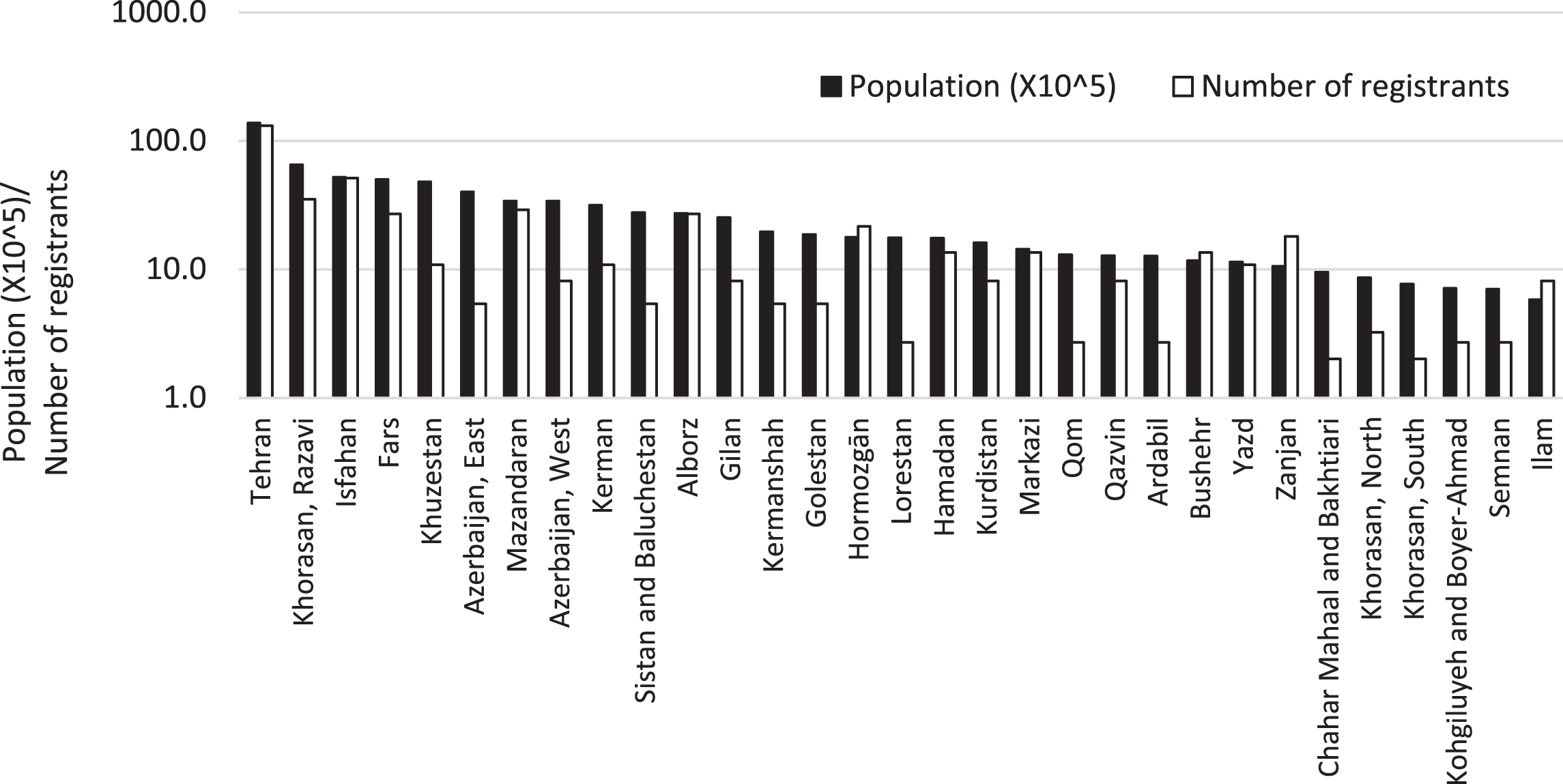
Population vs Number of registrants in provinces of Iran.
Registry technical design
RegHub Cloudware provides the technical structure of the online registry. The database system is currently customized to collect DMD/BMD clinical data. We designed the database for online use with a multi-level user interface - web server - database server. All received data is collected and stored on the central server in a database that works under the SQL system. This registry is accessible over the Internet with web browsers of Firefox or Google Chrome by a 128-bit SSL encrypted connection. The hierarchical access system governs access to the database, and the database administrator monitors access and attendance of patients and centers. All participating centers are entitled to data access equally. A specific username and password generated for each registrant, used for data entry or modification. A questionnaire form was designed to collect personal and mandatory data according to TREAT-NMD Global Network guidelines. The registry controls all the data with a copy saved in a central server. Each registrant has access to the cumulative data as well as his own. An online request is available for third-party inquiries, which the registry steering committee evaluates, and in case of approval, grants a report with no personal information included.
Data entry type, informed consent
The registry data entry is a combination of patient entry confirmed by specialist and clinician entry. We collect patient data according to the national and international law (Iran Patient Data Protection Act 2009/387956 and European Union Directive 95/46/EC). A written informed consent from the patient or his guardian explaining the purpose and use of the database is mandatory for all patients’ records. All data are safe in the registry’s server and a print form in the registry central office archive located in the Pasteur Institute of Iran. Personal information is not disclosed to third parties unless required by law with a patients’ or his guardian consent to use the medical information included in the registry. Registration is voluntary, with no commitment to the patient or the registry. All patients’ information is confidential and can be eliminated upon the registrant request; however, we have not yet received such a request.
Data collection form (Table 1)
Data collection form
The data collected is following the minimal data set specified by the TREAT-NMD [13]. This information includes; clinical assessment, serum CK level, muscle biopsy, and genetic information, which is a mandatory item. The form includes epidemiological data, motor function, cardiovascular activity, respiratory status, history of scoliosis, steroid therapy and diagnosis of DMD/BMD/IMD or carrier. The information is updated annually either through an online questionnaire sent to registrants or through a phone call made by our team. The online form is demonstrated in (Table 1).
RESULTS
Demographic distribution
The registry includes data of patients from the 31 provinces of Iran. Tehran, the capital, has the highest number of registrant with 131 cases (26%), followed by Isfahan (10%), Khorasan Razavi (7%), Mazandaran (6%), Fars (5%) and Alborz (5%). The 12 provinces have less than 1%of participants in the registry. The ratio of the number of registrants to the population varies in the range of 1.7 to 0.14 registrant in each 100,000 of the provinces’ population. Tehran is in the 7th rank among the provinces, with a ratio of 0.96. (Fig. 2.)
Age distribution
The age distribution profiles of the registrants are different between DMD and BMD patients. The DMD biggest age group is between 6 and 10 years old, followed by the age group between 11 and 15, which are collectively 65%of all the DMD patients in the registry. The biggest BMD age group is between 16 and 20 years old, followed by age group between 21 and 25 which collectively are 52%of the BMD patients. The average age of registrants in the DMD group is 13.4 years old, and BMD is 20.7. (Fig. 3).
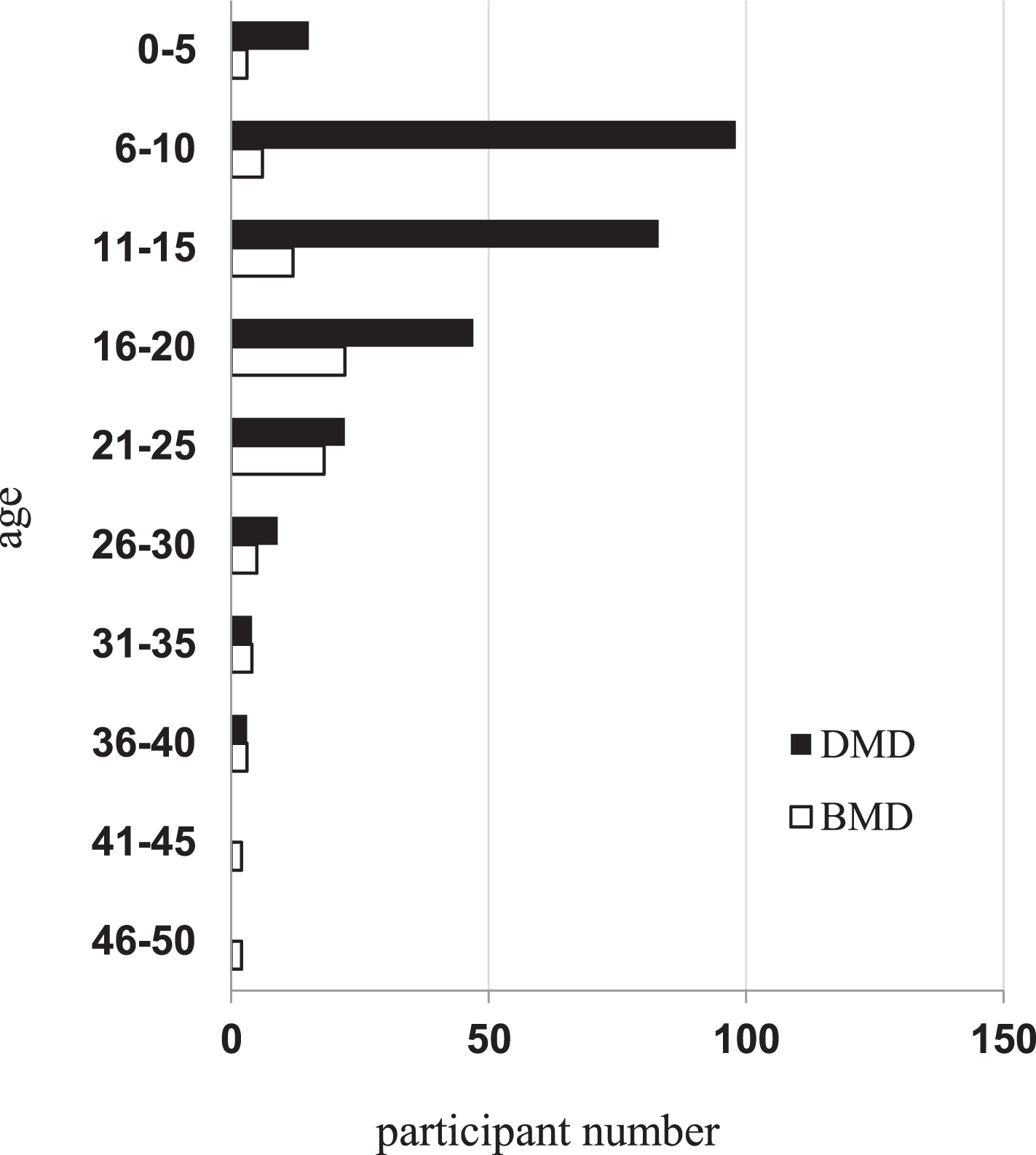
Age distribution of DMD/BMD registrants in the registry.
Diagnosis
As of March 2020, the dataset included 522 registrants; 281 DMD (56.1%), 77 BMD (15.4%), 7 (1.4%) with intermediate status, 104 carriers (20.8%) and 32 genetically unconfirmed dystrophinopathy patients with no confirmed mutation (6.4%). Genetic confirmation is either through multiplex PCR targeting the gene hot spots or/and MLPA for the whole 79 exons. 88%of diagnosed individuals with confirmed mutation used the PCR method in their diagnosis. Dystrophin gene sequencing is the method of choice if deletion or duplication is not found by other methods. Otherwise, a neuromuscular specialist will confirm the diagnosis based on the clinical assessment, elevated serum CK, and the lack of dystrophin in muscle biopsy. (Fig. 4).
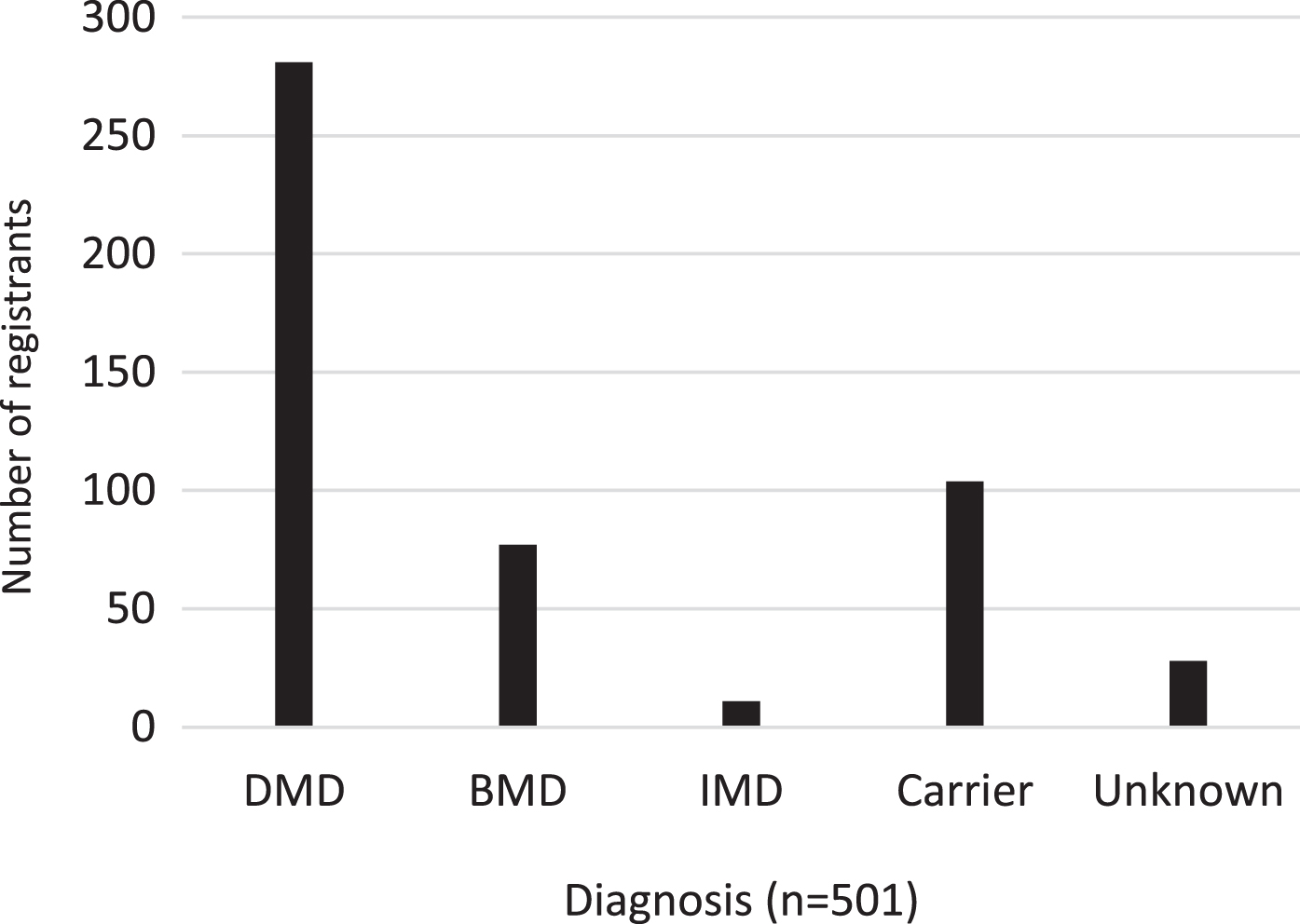
Dystrophinopathy diagnosis (n = 501).
Participants characterization (Table 2)
Number of registrant and their clinical symptoms (n, %)
Our team collected 92%of applicants’ data from data centers such as genetic labs, hospitals, research centers, or NGOs. In comparison, 8%of the data are entered by registrants directly after being approved by a neuromuscular specialist. 79.5%of registrants were actively monitoring their data by entering at least once into their profile or browsing registry’s general report pages. We collected the most common associated clinical information in each DMD/BMD group (Table 2).
93.4%of DMD and 52.0%of BMD patients had calf muscle hypertrophy. 89.2%of DMD and 39.7%of BMD patients presented with Gowers’ sign. The age of onset and severity differs between our DMD and BMD patients, where DMD patients showed a more severe presentation and early signs of cardiomyopathy. 14.2%of DMD and 6.8%of BMD patients showed signs of cardiomyopathy in the echocardiogram study. Regarding respiratory function (11%) of DMD and (2.7%) of BMD, patients used non-invasive ventilation or had low forced vital capacity in spirometry study. 3.6%of DMD and 2.6%of BMD patients had scoliosis. In DMD patients, only 22%and in BMD 11%were using steroids at least once. Moreover, 8.25%of our DMD/BMD patients had a positive family history of similar conditions.
Ambulation (Fig. 5)
We use the six-minute walk test to evaluate the ambulation status of DMD/BMD patients. We divide registrants into five groups according to their walking abilities. 45.2%of the DMD and 77.9%of the BMD registrants were able to walk more than 500 meters, 11.0%of DMD and 6.5%of BMD patients were able to walk 100–500 meters without any help, 5.3%of DMD and 5.2%of BMD patients could not walk more than 100 meters and 6.8%of DMD while 5.2%of BMD patients could only walk few meters. 31.7%of DMD and 5.2%of BMD patients were wheelchair dependent (Fig. 5). The average age for the loss of ambulation in DMD patients is 11.2 years old.
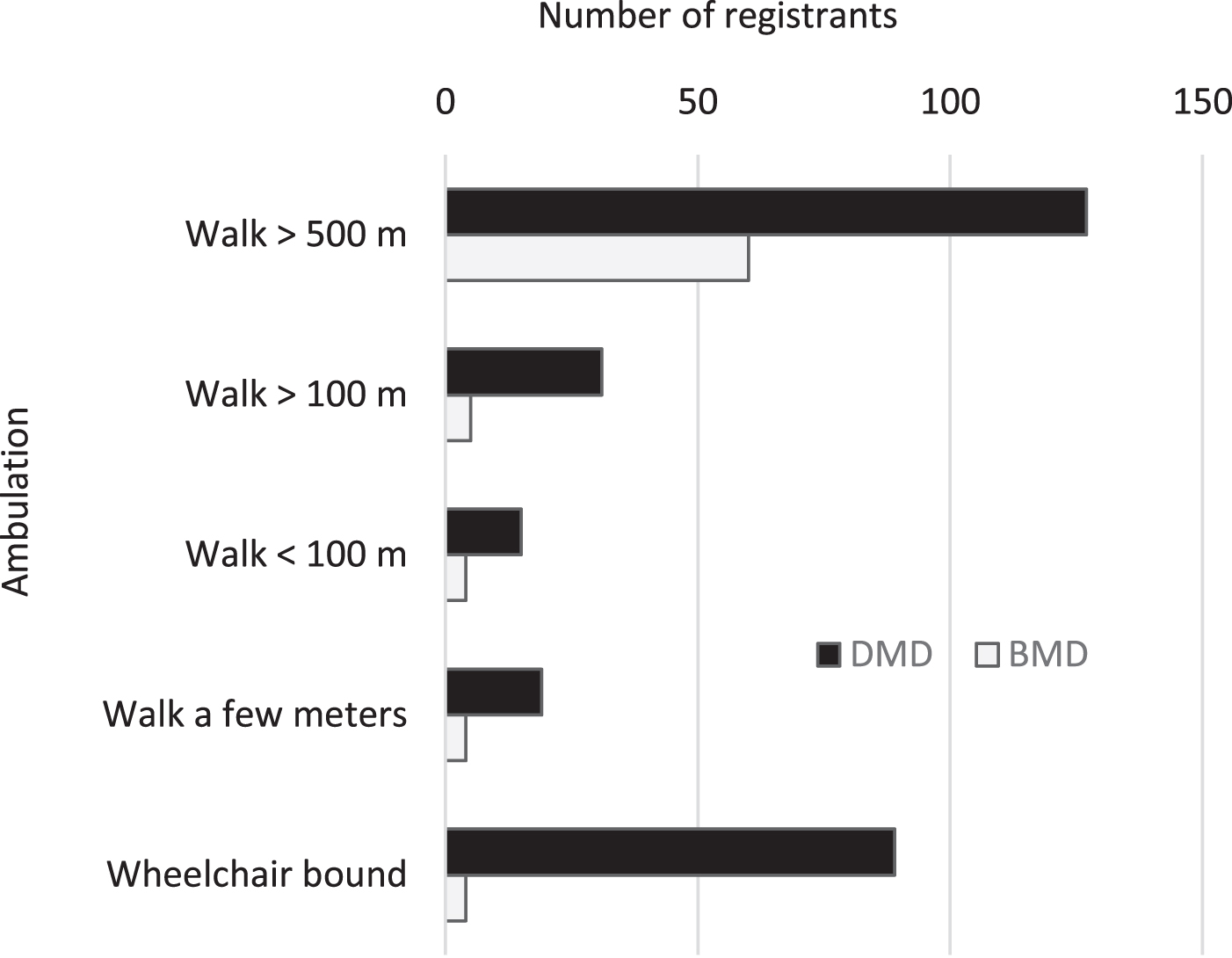
Ambulation status between registrants with DMD/BMD.
Mutation structure (Fig. 6 7)
In 358 DMD/BMD registrants, the most common genetic mutation was exon deletion (71.2%), followed by point mutation (13.7%), duplication (11.4%), and deletion/duplication (3.7%) (Fig. 6). The dystrophin gene two main hot spots are between exons 2–32 and 42–52. The average number of deleted exons in each registrant is 4.67; exon duplication is 12.95 exons, yet the duplication mutation was less frequent with no specific hot spot, unlike in the previous studies [14]. The most frequent exon deletion are in exons 50 (6.8%), 48 (6.4%), 46 (6.2%), 49 (6.2%), 47 (6.0%), 51 (6.0%) and 45 (6.0%). The frequency of the exon deletions and duplications in all the DMD/BMD registrants are demonstrated in (Fig.7).
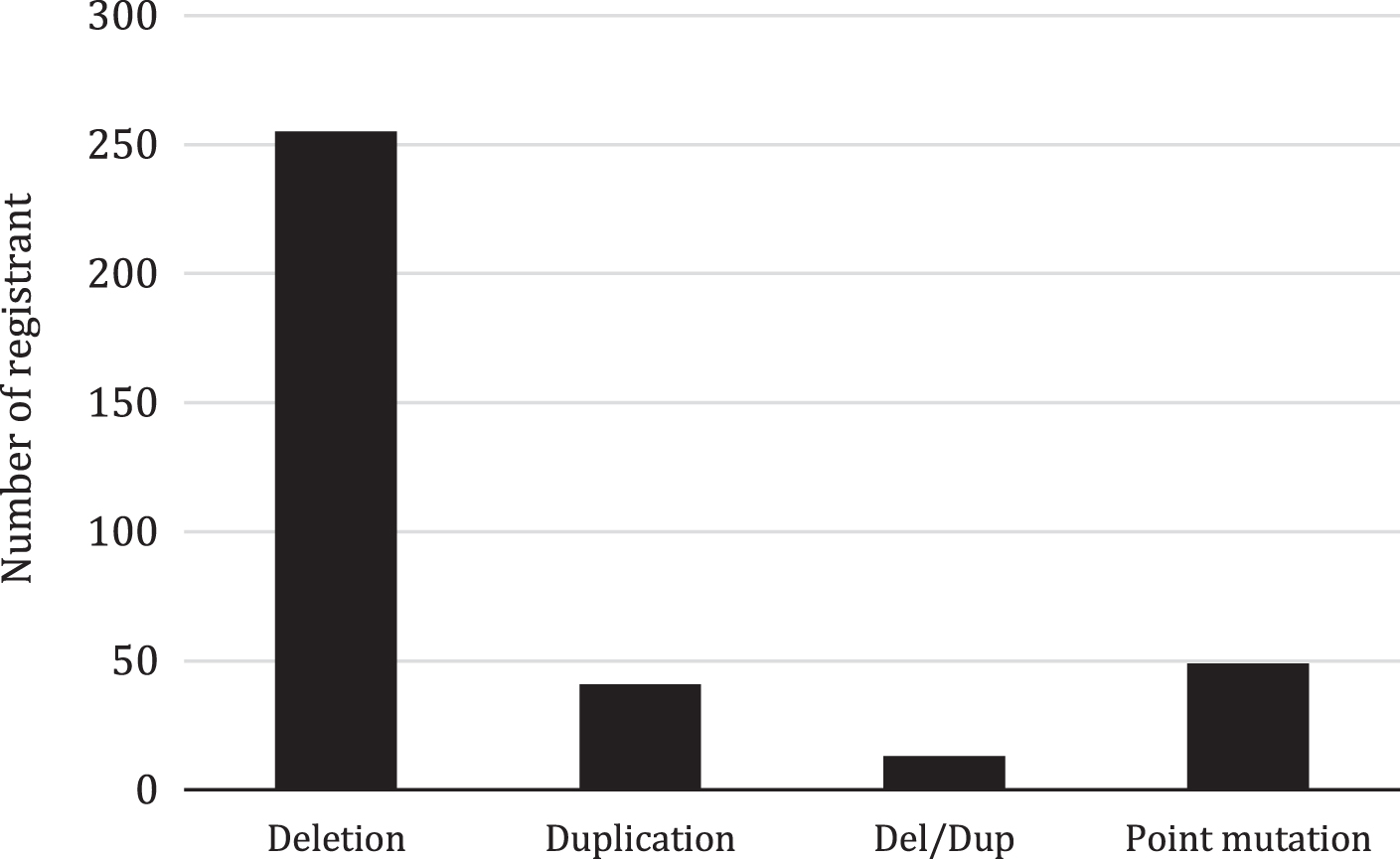
Mutation types in DMD/BMD registrants (n = 358).
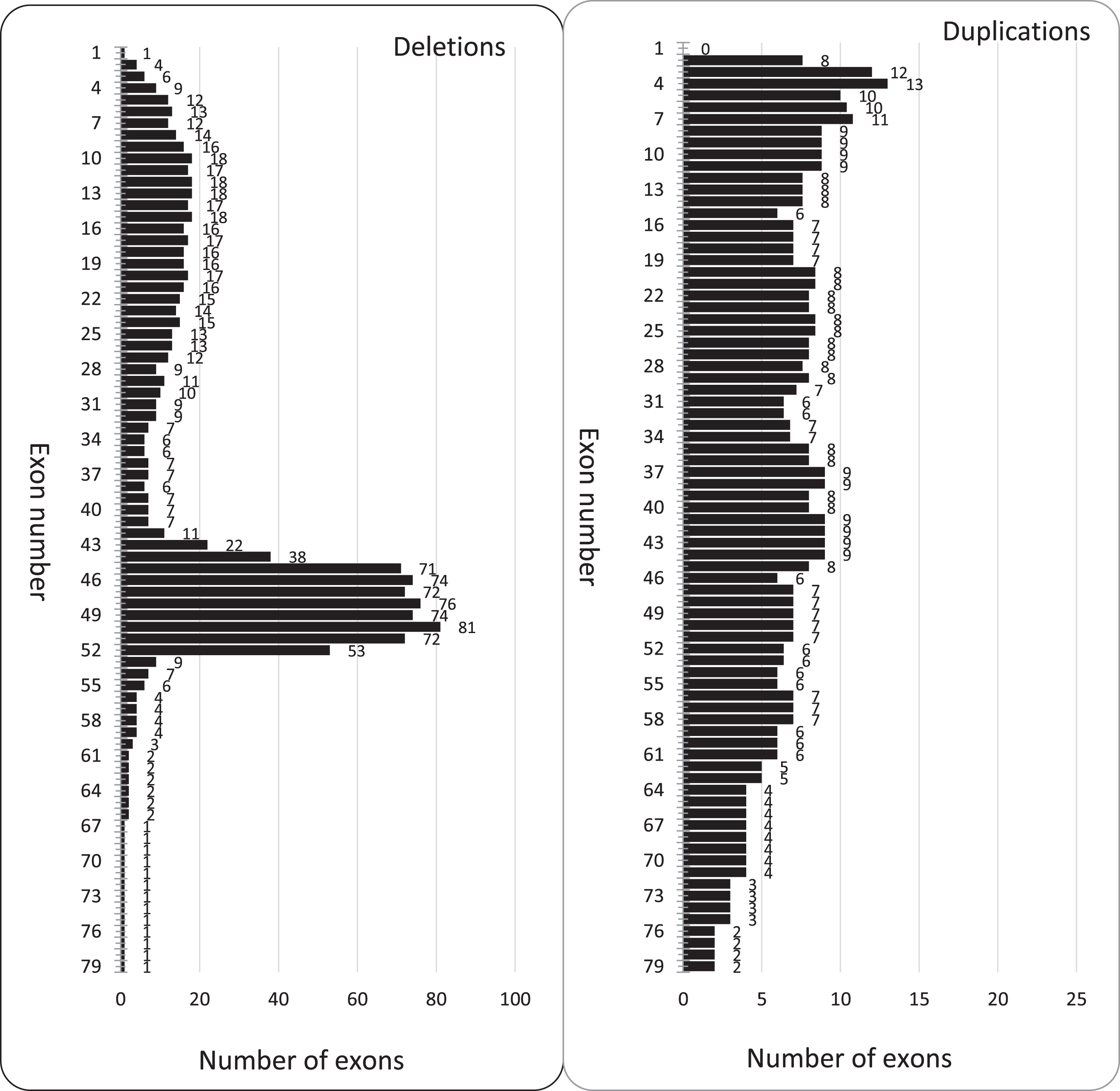
Deletion/Duplication of exons in DMD/BMD registrants.
DISCUSSION
The demographic distribution of our registry cases is not compatible with the population distribution in each province. Population to patient ratio should be equal for each province if the DMD/BMD distribution is uniform in all of the country, consistent with previous reports from other registries [15–17]. Currently, the leading laboratories and medical genetic data centers in some provinces are not yet well identified, and thus the registry does not fully cover the entire country. It is necessary to provide education and training for patients, families, caregivers, laboratory staff, and medical centers about the importance and benefits of data sharing. Networking and data sharing is mandatory to establish a national registry. All patients’ medical records referred to hospitals and medical centers in Iran are centralized in EHRS database. Connection to this database in order to obtain raw data and identifying possible new cases will make it possible to add new DMD/BMD cases to IRDAB which is in process. Small part of the data is entered into the registry by registrants. Centers or physicians supply the central part; however, the number of patients using online data access is relatively high, which is considered a good indicator of patients’ interest in their data and self-tracking. This high number is significant and highlights the registry’s success in this issue.
The ratio of deletions, duplications, and point mutations in our registry is in accordance with reports from other registries [18]. The results of our registry data analysis indicated that the severity of the disease in our patients depends on disruption of the dystrophin gene reading frame rather than on the range or extent of the exons deleted or duplicated fragments. The exon deletion distribution in the two “hot spots” regions between exons 2–32 and 42–52 is consistent with previous reports from other populations [19, 20].
The registry contains phenotype genotype profile of 358 DMD/BMD patients. The severity of symptoms, loss of ambulation, cardiomyopathy and patients on non-invasive ventilation is generally more in DMD patients compared with BMD cases in our registry.
The percentage of DMD patients at least once on steroids is relatively low (22%) in Iran compared to the number of patients in Europe or the United States. Reports show that the number of patients who had been at least once on steroids from the western European countries like Bulgaria (29%) or Czech (32%) is lower compared with the western European countries like Germany (73%) or the UK (84%) in a cohort of 1000 patients [21]. While in the United States, 75%in a cohort of 1500 was reported on steroids [22]. Potential reasons for such a low number of patients on steroids include the lack of patient compliance to management after their diagnosis, another important reason is the lack of consensus among physicians regarding the steroid therapy due to the side effects. Nevertheless, more physicians and families became aware of the importance of standards of care and steroid therapy to prolong the ambulation time and improve the patients’ quality of life, following the examples in the developed countries.
Iran’s registry has been linked to TREAT-NMD since 2015. TREAT-NMD helps coordinate and standardize the diagnostic and therapeutic measures and facilitate the research in neuromuscular disease. IRDAB has outstanding attention to the DMD/BMD carriers’ diagnosis by collecting their genetic data and working to standardize their access to genetic counseling and prenatal diagnosis.
In the Iranian population of 82 million, we estimate the number of affected people to be around 3700 patients; thus, only 13.5 percent of Iran’s affected population registered.
Further study and collecting extra data is necessary for several reasons. A complete registry with genetic data of DMD/BMD patients and carriers will be obligatory to implement the ongoing national program to control muscular dystrophies. This program aims to identify all patients and carriers, which is critical. The registry serves as a starting point for this program and will facilitate the prenatal diagnosis, newborn screening, treatment, and rehabilitation of people with DMD/BMD.
There is great potential in the scientific community and the country’s molecular medicine networks to initiate clinical trials and improve research on muscular dystrophies. IRDAB will have a significant role in providing sufficient and accurate molecular data of DMD/BMD individuals. One of these registry goals is to facilitate the easy and rapid participation of eligible patients in national/international clinical trials and outcome measures studies. The registry received six academic and pharmaceutical inquiries from TREAT-NMD and four inquiries from medical universities in the country.
CONCLUSION
The development of database infrastructure, networking, and education programs for patients and families are still essential. The registry helped to identify carriers that would reduce the DMD/BMD birth rate in our country and improve patient’s quality of life by guiding patients’ standards of care. Furthermore, registrants’ data will be used in the future to recruit patients for clinical trials.
FUNDING
This project has been funded by the Ministry of Health and medical education of Iran, under Grant No. 301/700, subject to 1%of Article 56.
CONFLICT OF INTEREST
None of the authors have any financial interest in this project or publication of this article.
Footnotes
ACKNOWLEDGMENTS
We would thank all the Duchenne and Becker patients and their families who have participated in this registry. Our appreciation to Dr. Craig Campbell, MD, Ph.D. Chair of TREAT-NMD Global Database Organization Committee and Dr. Rasha El Sherif MD, Ph.D. Chair of the TGDOC Publications Committee for their help, advice, and comments on the manuscript. We would like to thank the Pasteur Institute of Iran for its administrative support. The TREAT-NMD Global Neuromuscular Network supports this study.