
Abstract
Neuropathies in Myoclonic Epilepsy with Ragged Red Fibers (MERRF) syndrome are frequent but ganglionopathies have never been reported. We retrospectively identified 24 patients with MERRF mutations in the neuromuscular center Nord/Est/Ile de France (Pitié-Salpêtrière, Paris, France). Seventeen nerve conduction studies (NCS) were available. Five patients had MERRF syndrome and ganglionopathy, a pure sensory neuropathy. All of them displayed ataxia and mild clinical sensory abnormalities. Ganglionopathies have been reported in mitochondrial diseases but never in MERRF syndrome. We suggest that patients presenting with ganglionopathy, especially if associated with myopathy, lipomatosis or epilepsy, should be screened for MERRF mutations.
INTRODUCTION
Myoclonic Epilepsy with Ragged-Red Fibers (MERRF) syndrome was first described in 1980 as a multisystemic encephalomyopathy [1]. MERRF syndrome is due to a point mutation in mitochondrial DNA (most often m.A8344G) leading to a defect on the MT-TK gene encoding a lysine transfer RNA [2, 3]. MERRF is characterized by myoclonus, epilepsy, ataxia and patients may present myopathy, deafness, lipomas and cardiomyopathies [2, 4–6, 2, 4–6]. Peripheral neuropathies have been reported in MERRF syndromes with varying frequency (14.7% –49%) [2, 7]. Moreover, their nature is highly heterogeneous ranging from sensory or sensory-motor neuropathies with demyelinating, axonal or mixed features [8–11]. However, to our knowledge, ganglionopathy - a non-length dependent pure sensory neuronopathy affecting the dorsal root ganglia –has not been reported in MERRF mutations to date.
We describe five MERRF patients with classic m.A8344G mutations diagnosed with ganglionopathies during their follow-up in the Pitié-Salpêtrière hospital neuromuscular reference center.
METHODS
In a cohort of 24 patients presenting with MERRF syndrome (22 with m.A8344G, one with m.G8363A, one with m.T8355 mutations), we retrospectively analyzed 17 available nerve conduction studies (NCS) of patients followed at the neuromuscular reference center of the Institute of Myology in Pitié-Salpêtrière Hospital between 1996 and 2014. Data of patients diagnosed with sensory neuropathy or ganglionopathy were recorded. All patients had underwent clinical, biological, neurophysiological, cardiological and respiratory assessment.
MERRF diagnosis was established by muscle biopsy and mitochondrial DNA analysis. Histological and chemical studies on frozen muscle sections included specific analysis with Hematoxylin-eosin-saffron, Gomori’s trichromes, succinate deshydrogenase, oil-Red O and Cytochrome C oxidase stains from deltoid biopsies.
Mitochondrial DNA was extracted from muscle biopsy by standard procedure and from blood, buccal cells, urine and hair samples with a rapid extraction kit (Qiagen®). Mitochondrial tRNALys gene (MT-TK) screening was carried out by standard procedure as previously described [4].
Electromyography (EMG) and NCS were carried out for each patient either at diagnosis or during follow-up. If available, data on Sensory Nerve Action Potentials (SNAP) and Sensory Nerve Conduction Velocities (SNCV) were collected on radial, median, ulnar, superficial peroneal and/or sural nerves. Motor Nerve Conduction Velocities (MNCV), Distal Latencies, Compound Muscle Action Potentials (CMAP) were collected on median, ulnar, fibular and tibial nerves. Ganglionopathy was defined according to the electrophysiological criteria of Camdessanché et al, i.e., at least one absent or diminished (>30%) SNAP on three upper-limb nerves, and less than two abnormal motor nerve conduction [12]. Patients with ganglionopathy had been screened for other causes of sensory neuronopathy: vitamin B9, B12 and E levels, protein immunoelectrophoresis, ANA, anti-SSA-SSB, anti-transglutaminase, anti-ganglioside and anti-neuronal antibodies.
RESULTS
Five of the 24 MERRF patients in our cohort had ganglionopathies. In the remaining patients, seven did not have NCS data; four had normal NCS; one had axonal sensorimotor neuropathy; and seven had length-dependent axonal sensory neuropathies. Consequently, 13 of the 24 (54%) MERRF patients had proven neuropathies and five (21%) had ganglionopathies. In other terms, 38% of MERRF neuropathies were ganglionopathies with clinical score according to Camdessanché criteria of 9 (positive if score >6.5) for each patient [12]. Three of the five patients in the ganglionopathy group were male. The age ranged between 19 and 61 years and two were mother and son. They all had the classic m.A8344G mutation. The analysis of patients heteroplasmy rate from different tissues showed an average of 75.8% of mutated DNA in the blood (5/5 available data), 65% in buccal cells (4/5), 50% in hair (4/5), 79.3% in urine (3/5) and 77.6% in muscle (3/5). Details for hetreroplasmy rates in the different tissues are presented in Table 1. Mitochondrial abnormalities (Ragged Red Fibers and COX negative staining) were present on the five muscle biopsies. For all five patients, NCS showed absent or diminished SNAP on both upper and lower limbs without significant motor involvement. The mean SNAPs were 4.6 microvolt (μV) for median nerves, 2.6μV for ulnar nerves, 5μV for radial nerves, 2.9μV for superficial peroneal nerves and 2.5μV for sural nerves. All the NCS data are presented in Table 2. EMG showed either myopathic features or were normal. Neuropathic features were described in only one muscle for one patient. All patients displayed clinical sensory abnormalities (hypoesthesia, hypopallesthesia), which were restricted to the lower limbs. They presented ataxia with both a proprioceptive and a cerebellar component and deep tendon reflexes were decreased or absent in all cases. In our cohort, apart from less clinical sensory lower limb involvement (100% vs 57%), the seven patients with length-dependent sensory axonal neuropathies did not seem to differ from the ganglionopathy subgroup of patients in terms of classic MERRF features. 8/24 patients with MERRF were wheelchair bound (3/5 with ganglionopathies, 2/7 with sensory neuropathies). The clinical data of patients with ganglionopathies and those with length dependent sensory neuropathies are presented in Table 3.
Patients with ganglionopathies (heteroplasmy rate in different tissues are expressed in percentage)
NA = not available.
Nerve Conduction Studies for the 5 MERRF patients diagnosed with ganglionopathy
A = Ankle; CMAP = Compound Motor Action Potentials; L = Left; m/s=meters per second; mV = millivolt; NCS = Nerve Conduction Studies; NV = Normal Values; R = Right; SCV = Sensory Conduction Velocities; SNAP = Sensory Nerve Action Potentials; μV=microvolt; W = Wrist.
Clinical data from MERRF patients with sensory neuropathies and ganglionopathies
DISCUSSION
In our cohort of 24 patients with MERRF, 54% patients had overall neuropathies and among them five cases of ganglionopathy were identified out of the 17 available NCS. Ganglionopathies are pure sensory neuronopathies, corresponding to dorsal root ganglia disorders. Despite this definition, clinical differentiation with length-dependent axonal sensory neuropathies may be difficult. Therefore, Camdessanché et al proposed clinical and electrophysiological criteria to accurately diagnose ganglionopathies in 2009 [12]. While the five patients described in our study fitted these electrophysiological criteria, clinical features were more difficult to prove: all the patients presented ataxia but not upper limb sensory symptoms or asymmetric presentation. However, these clinical criteria would appear to be more appropriate for acquired rather than hereditary ganglionopathies since they rely on data extracted from cisplatine, dysimmune, paraneoplastic and idiopathic neuropathies. Nevertheless, ganglionopathies have also been reported in mitochondrial diseases such as SANDO syndrome (Sensory Ataxic Neuropathy, Dysarthria and Ophtalmoparesis) associated with POLG mutations [13]. Ganglionopathy is also a classic feature of CANVAS (ganglionopathy with gastroesophageal reflux, cough and ataxia) and Friedreich (ganglionopathy with pyramidal syndrome and ataxia) disease, the latter one being characterized by mitochondrial dysfunction through iron accumulation and free radical production [14, 15]. To the best of our knowledge, ganglionopathies have not been yet described with m.A8344G or other point mutations of MERRF syndrome. Neuropathies are reported as being frequent in MERRF patients and are mostly sensory and slowly progressive [10, 11, 16]. NCS and biopsy studies are heterogeneous and may reveal axonal or mixed axonal and demyelinating polyneuropathies [9, 19]. However, histological findings in mitochondriopathies including MERRF syndrome have shown that the dorsal root ganglia harbor degenerative features, supporting the idea that the pathophysiology of MERRF is consistent with the development of ganglionopathy [20]. There are previous reports on sensory ataxic neuropathies in patients with m.A8344G mutations but available data are insufficient to conclude that they fit the criteria of ganglionopathies [21, 22]. Interestingly, Luigetti et al noted in their study of MERRF patients that the mean radial SNAP amplitude was lower than the sural one, a feature that is consistent with ganglionopathy [7].
Despite the fact that MERRF syndrome was first described in 1980, cohorts with detailed electrophysiological data are lacking in this disease. Among our patients, seven had no NCS examination, probably because they were paucisymptomatic. In our cohort, the five patients with ganglionopathy were more often wheelchair bound compared to the 19 remaining patients. One explanation of severe ataxia could be the presence of additional ganglionopathy.
There are limitations in our work. This study was retrospective and some clinical data, especially concerning the sensory profile, and NCS data were missing in the overall MERRF cohort. While ataxia
was a consistent feature in our ganglionopathy cohort, the patients did not present asymmetric features or predominant upper limb sensory involvement. However, as mentioned above, these features are more likely to be characteristic of acquired than genetic ganglionopathies and similar observations can be made for SANDO or Friedreich diseases.
To conclude, the five cases reported here constitute the first description of MERRF patients displaying signs of ganglionopathy with strong confirmation on NCS. Whereas we know that the main feature in MERRF syndrome is ataxia, we should probably be aware that ataxia may not be of cerebellar origin alone, but could be of mixed origin and at least partially related to a ganglionopathy. Moreover, we suggest that patients presenting with ganglionopathy should be screened for mitochondrial mutations, and specifically MERRF disease, especially if associated with myopathy, lipomatosis or epilepsy (Fig. 1).
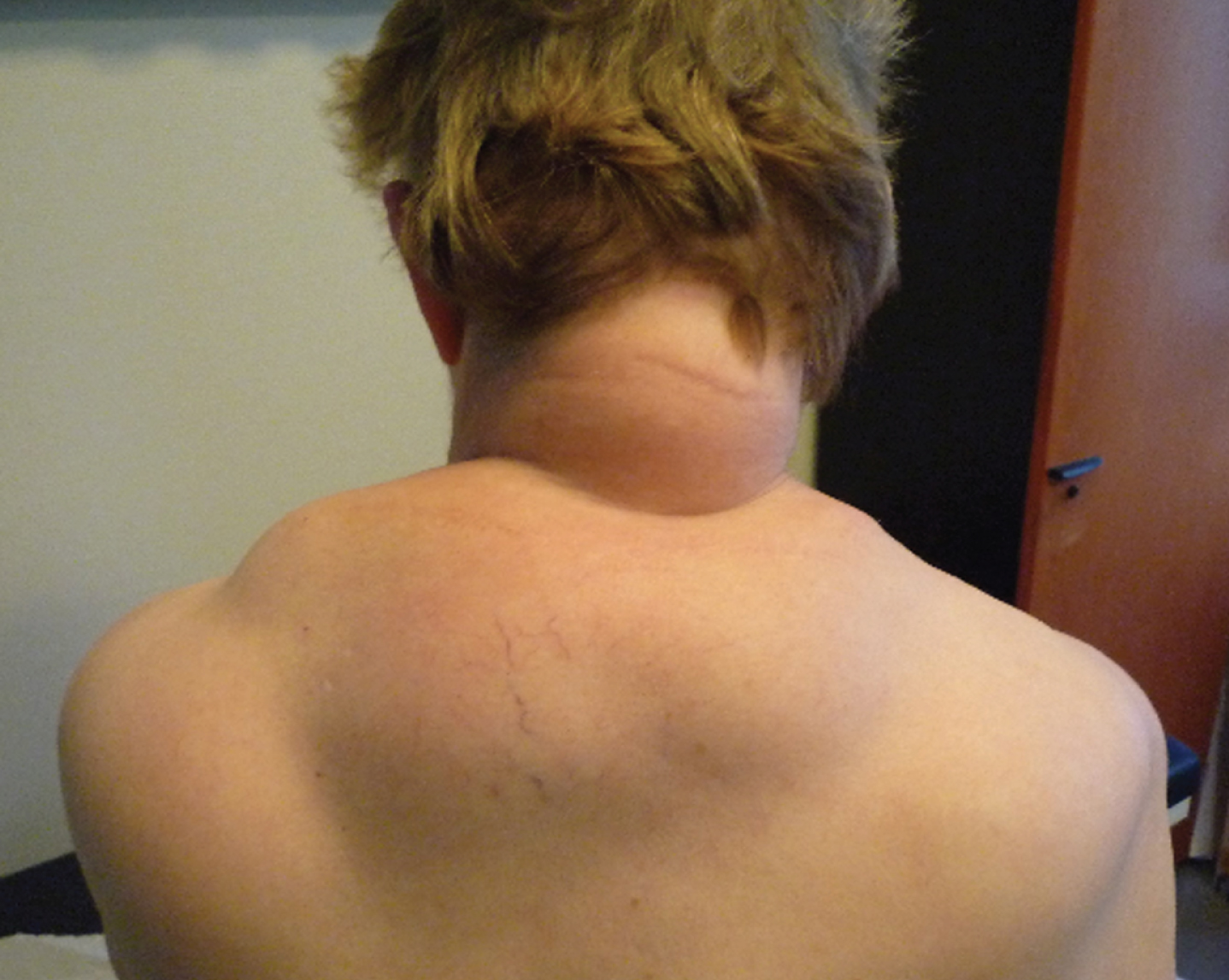
Patient with MERRF syndrome presenting a large lipoma (for the online PDF).
All authors have contributed to the work, agree with the presented findings. The work has not been published before nor is being considered for publication in another journal.
FUNDING SOURCE
This work was completed with no specific funding.
DISCLOSURE STATEMENT
The authors have no conflict of interest to report.
Footnotes
ACKNOWLEDGMENTS
None.