
Abstract
Spinal muscular atrophy (SMA) is a neuromuscular disorder affecting young children. While pre-clinical models of SMA show small spleens, the same is not true in humans. Here, we show by doppler ultrasonography decreased splenic blood flow in Smn2B/– mice. Further, AAV9-SMN gene therapy does not rescue the distal ear and tail necrosis nor the spleen size in these mice, suggesting that the latter may be linked to a cardiovascular defect. Absence of smaller spleens in human patients is likely due to differences in presentation of defects in SMA between pre-clinical mouse models and human patients, particularly the susceptibility to cardiovascular issues.
INTRODUCTION
Spinal muscular atrophy (SMA) is a devastating neurological disease, marked by paralysis and muscle wasting [1]. SMA is caused by deletion or mutation in the ubiquitously expressed Survival motor neuron 1 (SMN1) gene [2], essential for many RNA-mediated functions and splicing [3]. Interestingly, several reports identified non-neuronal defects in SMA [4, 5]. These include description from three groups on lymphoid organ defects in mouse models of SMA [6–8]. The most striking abnormality was the small size of the spleen in comparison to control animals [6–8]. In contrast, a small cohort of human patients showed some splenic defects of unknown significance, however, there was no mention of size difference [8]. Thus, it is not clear why the preclinical models consistently showed small spleens whilst human patients did not. The difference in spleen size in pre-clinical models could be due to abnormal vasculature, denervation and/or cell-intrinsic defects [9–11].
Nearly all SMA mouse models show distal tissue necrosis [12–17], a feature believed to be caused by poor perfusion due to vasculature defects [11, 19]. In fact, the necrotic tails in the mild C/C model of SMA displayed loss of intact lateral vasculature due to vessel wall abnormalities [16]. This was preceded by a significant decrease on thermal imaging, indicating poor blood supply [16]. It was suggested that the necrosis could be secondary to poor vasculature [16]. Similarly, the evaluation of tail necrosis revealed thrombosed small blood vessels and fibrinoid necrosis within the vessel wall [18]. In contrast, there are only five case reports of vascular problems in SMA patients, presenting with distal necrosis [20–22]. It is also noteworthy that the spleen size in a mouse model of SMA is initially normal at birth and gradually diminishes after birth [6]. This suggests that the transition from fetal to neonatal circulation may be unable to sustain the volume of the spleen [6]. In line with this, SMN depleted hearts show reduced function as exemplified by low cardiac output, bradycardia and heart block in preclinical models [10, 23–25]. Structural and rhythm defects have been suggested to be present in SMA patients, but do not appear to be present in all SMA patients [22, 27]. Considering our current knowledge, the defects in heart and vasculature (hence poor perfusion capacity) [28] appear to be the most likely reason for the reduced spleen size. Physiologically, it should be noted that the spleen is highly vascularized and acts as a large reservoir of blood containing up to 10–50% of the red blood cells depending on the organism [29, 30].
Here, we further explore in Smn2B/– mice defects related to perfusion of lymphoid organs, both in the natural course of the disease and following AAV9-SMN gene therapy. We show by doppler ultrasonography that Smn2B/– mice have lower perfusion capacity with lower systolic peak flow velocity in their splenic artery prior to major changes in spleen size. We also show that AAV9-SMN gene therapy does not rescue the size of the spleen nor the distal necrosis. However, it partially restores the histological architecture in the spleen. In contrast, the thymus, another lymphoid organ, shows normal size and architecture after AAV9-SMN treatment. We conclude that the diminished splenic size is consequent to poor perfusion in SMA preclinical models, a feature that is rare in SMA patients.
MATERIALS AND METHODS
Mouse models
The Smn2B/– (C57BL/6J background) [31] mice were housed at the University of Ottawa and cared for according to the Canadian Council on Animal Care (CCAC). All procedures involving experimentation on animal subjects are done in accord with the CCAC. Birth was designated as P0.
Ultrasonography
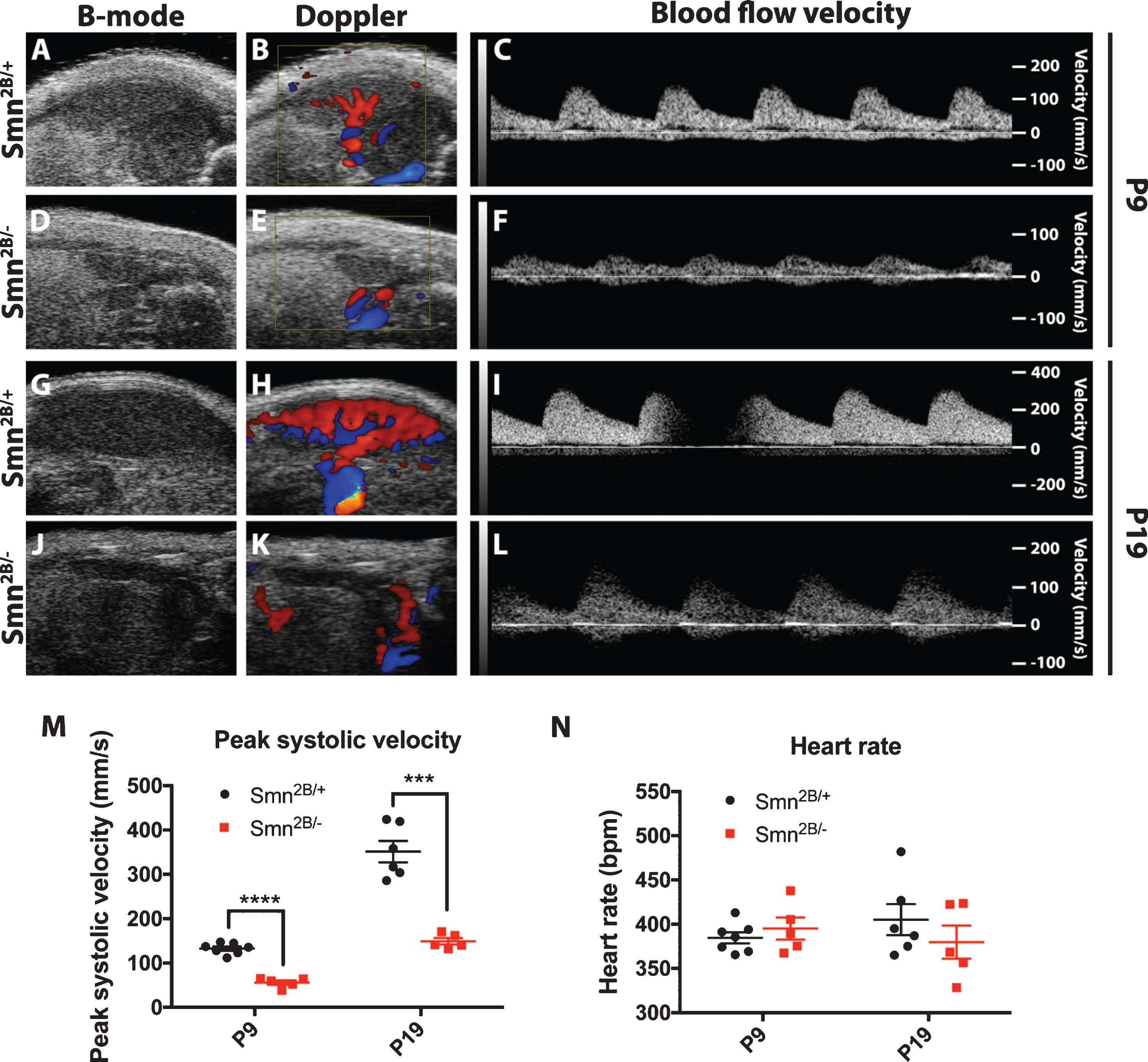
Mice were shaved and Veet was used to ensure complete hair removal, allowing for adequate ultrasonographic quality. Mice were anesthetized using 1–2% continuous isoflurane and placed on a warming movable station where each limb was taped to the station to allow heart rate monitoring. For P9 mice, the isoflurane pipe was extended and taped to the head of the mouse to ensure proper anesthetic delivery given their small size. Ultrasonography was performed using a MS400 or MS550 transducer and the VEVO 2100 (FujiFilm VisualSonics, Toronto, Canada). The spleen was identified under B-mode (2-dimensional ultrasound image). Doppler was used to identify the splenic artery and to measure blood flow. Peak flow velocity was sampled as recommended by FujiFilm VisualSonics consultant. Mice were repositioned in a right lateral decubitus when visualization of the splenic anatomy and vascularity was difficult. At least 3 measures of systolic peak flow velocity were measured and averaged. Heart rate measurement was obtained through the distance between each systolic velocity peak.
AAV9-SMN production and injection
The self-complementary scAAV9-SMN vector was produced by calcium phosphate transfection of HEK293-AAV cells (Agilent) with pAAV-CB-SMN [32] and pDF9 plasmids. Briefly, the vector was purified from the cell lysate using a iodixanol density gradient followed by anion exchange chromatography (HiTrap Q-FF column, GE Healthcare). The scAAV9-SMN vector was finally resuspended and concentrated in DPBS on a centrifugal filter unit (Amicon® Ultra-15, Millipore). The titers of the vector suspensions were determined by qPCR using an amplicon located in the inverted terminal repeats as described in [33]. The obtained titers of the scAAV9-SMN vectors were 9.6×1013 VG/mL and 3.0×1013 VG/mL. Smn2B/– and Smn2B/+ mice were injected with 5× 1010 AAV9-SMN viral vectors at P1 through the facial vein. The mice were left to age until P19 for further analysis.
Gross morphology and histology
Morphological and histological observations were made as described previously [6].
Statistics
Data are presented as the mean±standard error of the mean. A two-sided Student’s t test was performed using Microsoft Excel or GraphPad Prism to compare the means of data when only two groups were compared (i.e. wild type vs. Smn2B/–). One-way ANOVA with Tukey’s multiple comparison test was used when multiple groups were compared. Statistical analysis used are described in each figure legend. Significance was set at P≤0.05 for *, P≤0.01 for **, P≤0.001 for *** and P≤0.0001 for ****. The N number for each experiment is as indicated in the figure legends.
Data and materials availability
Raw data can be provided upon request.
RESULTS
To measure blood flow to the spleen, we employed an in vivo approach by using doppler ultrasonography of the splenic artery in Smn2B/– mice. This is a superior technique to assess aberrations in splenic perfusion as it provides a real-time physiological reading of the perfusion of the spleen in comparison to qualitative assessment of blood vessels by histology. The splenic artery blood flow during the systolic phase was significantly reduced in Smn2B/– mice in comparison to control littermates at P19 (Fig. 1G–M). We next wanted to see whether this reduction in peak blood flow velocity was present at earlier time points, such as P9, where the size difference in the spleen is much less obvious [6]. This could represent an early pathogenic event, indicating that reduced blood flow may be present earlier than spleen size reduction. Indeed, we observed that blood flow was consistently reduced at P9 in Smn2B/– mice during the systolic phase (Fig. 1A–F, M). As anesthesia is necessary for this procedure, it is possible that cardiovascular rhythm depression might be influencing blood flow in Smn2B/– mice. Interestingly, these differences could not be attributed to a heart rate difference as both groups displayed similar heart rates despite anesthesia (Fig. 1N).
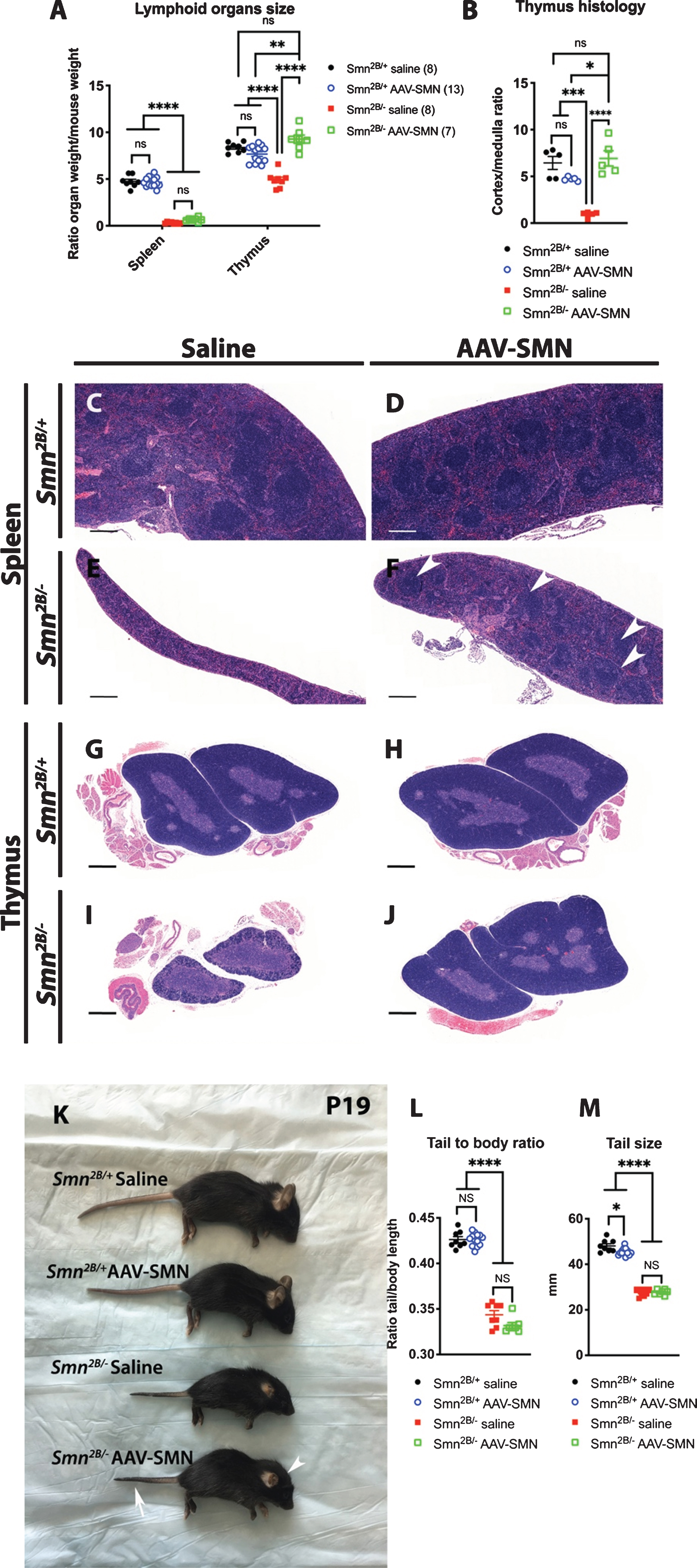
Next, we attempted to rescue spleen size by systemic viral mediated expression of SMN. Interestingly, similar gene therapy approach previously led to very modest improvement in heart functions [23, 24]. P1 Smn2B/+ and Smn2B/– mice were injected with AAV9-SMN via the facial vein and the spleen was extracted at P19 for morphologic and histological analyses. Our assessment revealed that AAV9-SMN administration did not increase the size of the spleen but did improve its histological structure (Fig. 2A, C–F). Noteworthy, the presence of white pulp was noticeable upon AAV-SMN treatment, a feature completely absent in untreated Smn2B/– mice (Fig. 2C–F). By comparison, the size of the thymus, which is also smaller in Smn2B/– mice, was completely rescued (Fig. 2A). The histological architecture, more specifically the cortex to medulla ratio, of the thymus was also rescued by AAV9-SMN (Fig. 2B,G–J). Interestingly, the necrosis of distal tissues in the ear and the tail of Smn2B/– mice, thought to be consequent of poor perfusion [16, 18], was not rescued (see arrow and arrowhead in Fig. 2K and quantified in Fig. 2L, M). This likely indicates that AAV9-SMN gene therapy failed to rescue defects related to blood circulation.
DISCUSSION
The identification of lymphoid organ abnormalities in pre-clinical models has raised the possibility that the immune system may play a role in SMA pathology. However, the most prominent finding, namely the drastically diminished spleen size in SMA mice, was not reported in a small cohort of SMA patients [8]. This is likely due to the inter-species differences in how SMN depletion affects various systems. However, no mechanism had been put forward to explain this difference.
In our study, we show that the small spleen size in mouse models of SMA may be a consequence of an increased susceptibility of mice to cardiovascular/perfusion defects upon SMN depletion in comparison to human patients. More specifically, we show that peak systolic velocity of blood flow in the splenic artery in Smn2B/– mice is diminished as measured by doppler imaging at all time points observed. Given that the spleen is mostly composed of blood circulating through its sinuses, it is perhaps not surprising that, much like a balloon being inflated, it is not reaching maximal size due to poor perfusion capacity. Indeed, this mechanism of action underlying spleen size is plausible for various reasons. The normal anatomy and physiology of the spleen, carrying a large amount of blood within it [29, 30], makes it susceptible to perfusion defects. The spleen size in Smn2B/– mice gradually becomes smaller postnatally [6] once it starts relying on neonatal circulation instead of fetal circulation. Many SMA mouse models display reduced cardiac function [10, 24], which could lead to poor perfusion. Moreover, systemic AAV9-SMN gene therapy did not yield much improvement for both the size of the spleen or the distal necrosis of ear and tail tissue, a process also thought to be mediated by poor perfusion [16, 18]. Significant residual cardiac impairment remains present after systemic AAV9-SMN gene therapy [23, 24], in line with our findings. This is likely because facial vein delivery of the viral vector does not reach or transduce adequately in all the tissues in the mice. Surprisingly, systemic AAV9 administration displays high expression in the heart [34], yet likely insufficient for SMN-depleted hearts, especially if damage had already incurred [23, 24]. Given that the histology of both the spleen and the thymus are improved, it is appropriate to assume that SMN reaches these target tissues. However, it is unclear whether it would efficiently target the nerves (despite AAV9 having tropism for the central nervous system [35]) or smooth muscles of the blood vessels [34, 37]. Furthermore, vascular issues have been identified in various organs [11, 24], including the spleen [6]. Lastly, mouse models of SMA display distal necrosis (short tails, smaller ears, loss of digits) [12–17], while only five case reports have described vascular defects in mostly severe SMA patients [20–22]. Given these results, the inefficient blood supply to the spleen is highly likely the reason behind its small size in the mouse models, and the reason why spleen size is not affected in human patients. Nevertheless, it is possible that alternative mechanisms such as abnormal proliferation, cell death, innervation, cell intrinsic defects and impaired homing chemokines contribute to this. There were mixed results on the proliferation in the spleen [7, 8], while cell death was not obvious [8]. An impact on the innervation of organs other than the skeletal muscle have been reported [10] but there are currently no studies on splenic innervation. Homing chemokines, serving as signals for immune cells to populate lymphoid organs and to organize properly, could be deficient [38]. Similarly, studies on intrinsic SMN-depleted immune cell defects have not been performed. It should be noted that impaired perfusion is likely not a contributor to all the splenic defects seen in SMA pre-clinical models. Indeed, histological architecture of both the thymus and the spleen were improved by systemic AAV9-SMN administration, which may point to cell-intrinsic defects underlying this aspect.
Overall, we provide evidence that poor perfusion is a likely contributor to small spleens in SMA mouse models and may explain the difference between SMA pre-clinical models and SMA patients.
CONFLICT OF INTEREST STATEMENT
Marc-Olivier Deguise received honoraria and travel accommodations by Biogen for the SMA Summit 2018 held in Montreal and the SMA academy 2019 held in Toronto, Canada. RK and the Ottawa Hospital Research Institute have a licensing agreement with Biogen for the Smn2B/– mouse model. These COI are outside the scope of this study. All other authors have no competing interests to declare.
AUTHOR CONTRIBUTION
MOD designed study, performed experiments, main contributor of the data, analyzed data, created all figures and wrote the manuscript. AB provided support for experiments. BS provided AAV9-SMN vector. RK designed study and prepared and wrote the manuscript.
Footnotes
ACKNOWLEDGMENTS
We would like to extend our gratitude to Sabrina Gagnon, Animal Care and Veterinary Services and the pre-clinical imaging core facilities (Gregory Cron) of the University of Ottawa, as well as Frederick R. Roberts from FujiFilm VisualSonics for assistance with experiments. RK was supported by Cure SMA/Cure SMA Canada; Muscular Dystrophy Association (USA) (grant number 575466); and Canadian Institutes of Health Research (CIHR) (grant number PJT-156379). BLS was supported by ERANET E-Rare FaSMALS (grant number 3ER30_160673). MOD was supported by Frederick Banting and Charles Best CIHR Doctoral Research Award.