
Abstract
Carey-Fineman-Ziter syndrome is a congenital myopathy associated with mutations in the MYMK gene. It is clinically defined by the combination of hypotonia, Moebius-Robin sequence, facial anomalies and motor delay. Historically it was considered a brainstem dysgenesis syndrome. We provide detailed information of a Spanish boy with compound heterozygous variants in MYMK gene. A muscle biopsy performed as a toddler only disclosed minimal changes, but muscle MRI showed severe fatty infiltration of gluteus muscles and to a lesser extent in adductors magnus, sartorius and soleus muscles. Clinical course is fairly static, but the identification of new well characterized genetic cases will help to delineate the complete phenotype.
INTRODUCTION
Carey-Fineman-Ziter syndrome (CFZS) was first described in 1981 in a pair of siblings with a distinctive pattern of malformation [1]. It is clinically defined by the combination of hypotonia, Moebius sequence, Robin sequence, facial anomalies, motor delay and growth failure. The aetiology has remained elusive, but historically some authors considered it was a brainstem dysgenesis syndrome [2, 3]. Recently CFZS (OMIM #254940) was found to be a congenital myopathy associated with autosomal recessive mutations in MYMK gene (previous symbol TMEM8C transmembrane protein 8C) (OMIM *615345), which encodes a muscle-specific membrane protein, named myomaker [4]. This protein controls myoblast fusion, a crucial step in muscle formation [5]. Only 12 genetically confirmed cases have been reported so far [4, 7]. We present an additional Spanish patient with two compound heterozygous mutations in MYMK. This is the first reported case caused by a splicing variant. So far, only missense variants have been reported. Our aim is to detail the clinical phenotype and highlight the diagnostic clues.
CASE REPORT
The patient is the first boy from non-consanguineous parents. His mother, who has hyperlaxity, is Spanish and his father Romanian. He has a healthy younger sister. During pregnancy a chorionic villus sampling was performed due to the presence of pharyngeal cyst hygroma. Chromosomal analysis revealed 46 XY karyotype and the hygroma resolved subsequently. The boy was born at 39 weeks of gestation without complications. His birth weight was 2.750 g, below the 10th percentile for male Spanish neonates, but his length was 51 cm and head circumference was 36 cm, both within the standard range (between 10th–90th percentile). At birth he was noted to be floppy and presented some dysmorphic craniofacial features which included small nose, retromicrognathia, downslanting palpebral fissures, low-set ears and a high arched palate. There was bilateral facial palsy without external ophthalmoplegia. Generalized joint laxity was evident, as well as global muscular hypoplasia. He also had feeding problems and needed nasogastric tube for two months. During the first years of life his motor development was delayed. He was able to control his head at 4 months and sit without support at 11 months. He also could walk independently around 2 years of age. At that time there was a striking axial hypotonia and most joints were hypermobile. He had mild dysphagia, difficulties with chewing and growth delay was evident. He also had camptodactyly and retractile testicles. Proximal limbs showed mild weakness and he used the Gowers’ maneuver to come to an upright position. He walked with a waddling gait and slight hyperlordosis. His comprehensive language and social skills were developing properly, although he had articulation difficulties due to the facial weakness.
A congenital myopathy with prominent facial involvement was suspected and several tests were performed. Brainstem dysgenesis was also included in the differential diagnosis. CK levels, nerve conduction study and electromyography of deltoid and quadriceps muscles were normal. At 3 years of age, cranial MRI did not show any brainstem abnormalities, but a muscular MRI disclosed moderate to severe fatty infiltration of gluteus muscles and mild involvement of adductors, sartorius and soleus muscles (Fig. 1). Nevertheless, at 18 months of age a muscle biopsy from quadriceps showed only minimal changes, with a few atrophic, predominantly type 2 fibers (Fig 2). A targeted gene sequencing panel which included 64 genes associated with myopathies was negative. FSHD1 and FSHD2 were genetically excluded. Finally, genetic testing by whole-exome-sequencing (WES) detected two variants in the MYMK gene. The missense variant c.271C>A (p.Pro91Thr) located in exon 3 and the donor splice variant c.399 + 5G>A located in intron 3. Sanger sequencing segregation studies demonstrated that both variants were inherited from his father and mother, respectively and absent on her unaffected younger sister. The missense variant (c.271C>A) previously associated with CFZS [4] has been categorized as a pathogenic variant. The splicing variant (c.399 + 5G>A) has not been previously reported. This is a rare variant on general population with a MAF < 0.0001. The bioinformatic analysis with the Alamut splicing package (Alamut visual 2.14) predicted that this variant alters splicing. To the best of our knowledge, this is the first reported splicing variant. Mutations at the canonical splice sequences usually lead to single exon skipping. Alternatively, this can also lead to the inclusion of an intron fragment. These changes, in the absence of functional studies, are predicted to result in the production of a nonfunctional protein. Nowadays he is 4 years old. His length is 102 cm (at the 30th percentile line) and his weight is 13 Kg (at the 5th percentile line). He has no cognitive impairment and his motor abilities are improving slowly. Axial hypotonia persists, but his speech and strength have ameliorated. However, he still needs moist and soft-textured foods that are easy to chew. He’s able to run and does not ‘climb up’ his thighs to get off the floor. His electrocardiogram and echocardiogram are normal and there is no respiratory involvement based on clinical exam.
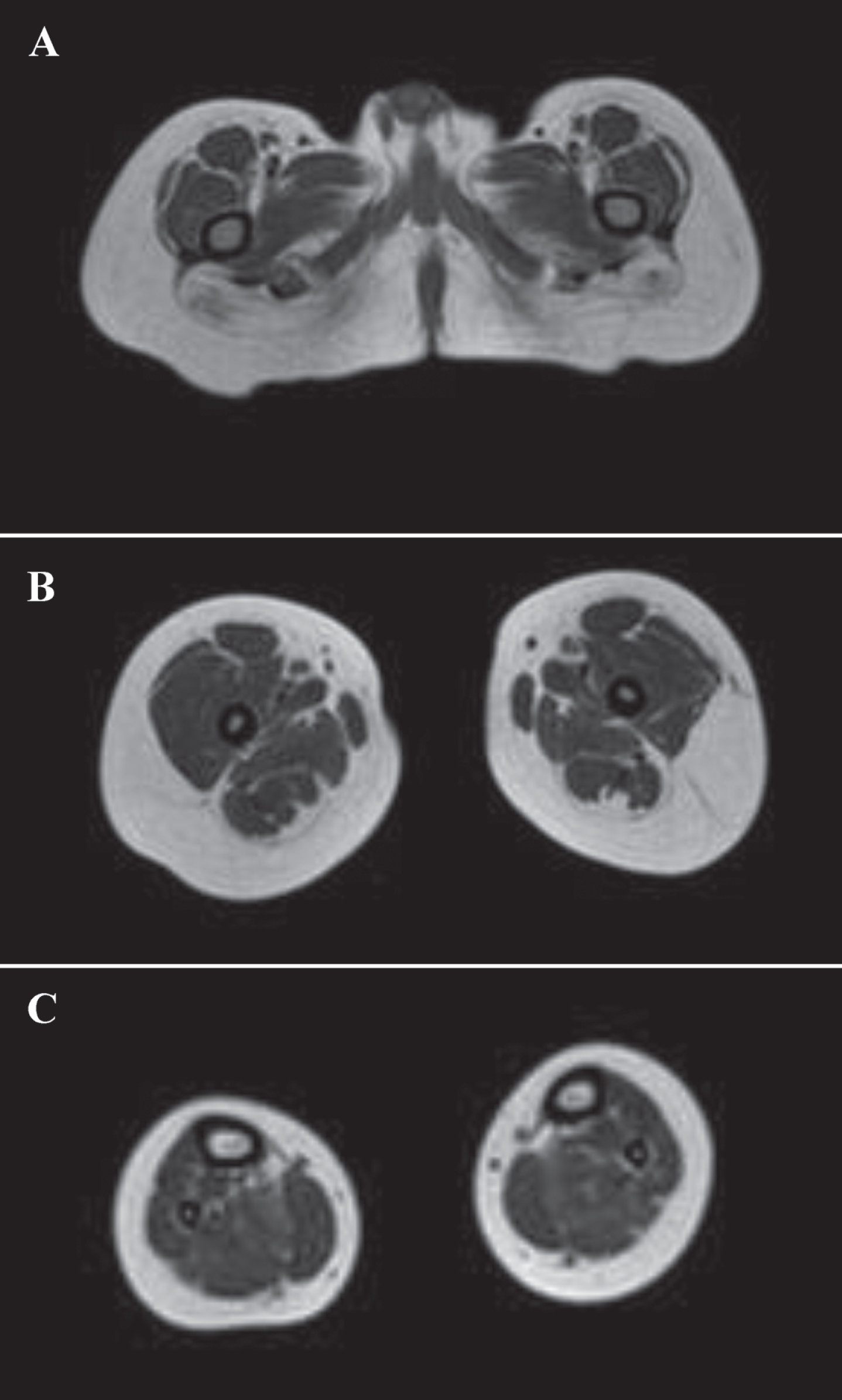
Muscle MRI showing moderate to severe fatty infiltration of gluteus muscles at thigh level (A), and mild involvement of adductors magnus, sartorius (B) and soleus muscles (C).

Quadriceps muscle biopsy showing no clear myopathic features (H&E staining, x200) (A), neither significant fiber type disproportion. There are very few scattered type 2 fibers which appear mildly atrophic (ATPase pH 9,4 ×200) (B). Bar = 50μm.
DISCUSSION
In this report we present a new patient with compound heterozygous variants in MYMK gene. He discloses the typical CFZS features: facial dysmorphism with Robin sequence, facial weakness and global hypotonia with motor delay. Although CFZS resembles a brainstem dysgenesis, it is an autosomal recessive congenital myopathy with prominent facial involvement. This disorder has been postulated to be caused by the dysfunction rather than elimination of the myomaker protein, as no loss of function variants have been previously reported. However, three of the previous missense variants have been shown to be functionally null variants. Myomaker is a fusogenic protein specifically expressed during muscle development. It induces fusion of mononuclear myoblasts to multinucleate myocytes in the skeletal muscle, and it is also essential in muscle regeneration [5, 8]. Multinucleate myofibers confer distinct biomechanical advantages to mature muscle by transducing force between remote skeletal attachments [4]. The molecular regulation of myoblast fusion is conserved during evolution. Knockout of MYMK in mice and zebrafish results in defective myoblast fusion, while heterozygotes are indistinguishable from wild type species [5, 9]. In addition, the homozygous mutant mice show early postnatal lethality. By contrast, the mutant zebrafish may survive, although the adult fish are smaller in size and develop cranial deformities resembling the facial defect observed in patients with CFZS [4]. CFZS pathophysiology is closely related to other myopathies caused by genes that are involved in skeletal muscle growth and regeneration, such as MEGF10 and PAX7, that specifically affect satellite cells. MEGF10 mutations reduce myocyte proliferation, differentiation, and fusion resulting in early onset myopathy, areflexia, respiratory distress, and dysphagia (EMARDD) [10], while PAX7 mutations impair satellite cells survival causing a disorder characterized by hypotonia, ptosis, muscular atrophy, scoliosis, and mildly dysmorphic facial features [11].
Before its genetic basis was discovered, some authors classified CFZS under the generic term of ‘brain dysgenesis’ [12] due to the occurrence of Moebius-Robin-Poland sequence at a variable degree. A few patients had additional brain features, such as cognitive impairment or cerebral malformation [13–15]. For De Giogia et al. many of these cases likely represent different disease entities [4]. The genetically confirmed cases share the characteristic facial appearance, which is the hallmark of the disease [4, 7]. All show facial weakness without abducens nerve palsy (or minimal limitations of eye movement in extreme positions of gaze). Other usual features are congenital hypotonia, generalized muscle hypoplasia and static or slowly progressive clinical course. Contractures, cleft palate as well as cryptorchidism in males are common. The phenotype resembles another congenital myopathy, originally described in Native American population, related to STAC3 mutations, although susceptibility to malignant hyperthermia is absent in CFZS [16]. Cardiac involvement has not been described either, but restrictive lung disease may develop in adulthood. Regarding complementary tests, CK levels are normal or mildly elevated. Muscle MRI has been performed in five adult patients in addition to our case. In general, there is fatty infiltration predominantly affecting the adductor magnus and sartorius muscles, and to a lesser degree the paraspinal and glutei maximi muscles [4, 7]. Lower-limb muscle MRI in our patient was consistent with that pattern, but there was severe fatty infiltration of gluteus muscles early in the course the disease. This is a relevant finding as he is the youngest patient reported so far, thus muscle MRI will be a useful tool for monitoring disease progression. Unlike the patient described by Alrohaif et al. [16], the boy did not present asymmetrical muscle involvement.
Histological analysis in MYMK mutant zebrafish reveals smaller single-nucleated myofibers and increased intramuscular adipocyte infiltration in skeletal muscles [9]. In patients with MYMK mutations the morphological information is scarce due to the paucity of muscle biopsy samples collected up to now. The most consistent pattern is either hypertrophy of both type 1 and type 2 muscle fibers –described in two patients [6]- or fiber type disproportion –described in one of the original siblings published in 1982 [1, 4]. Mild myopathic changes with internalized nuclei have also been reported in one adult [7]. However, in our case the muscle biopsy only showed a few scattered atrophic type 2 fibers, which expands the histopathological phenotype. One may postulate that the histological findings are influenced by the muscle selected for biopsy, since quadriceps was well preserved on muscle MRI. Another explanation would be the patient’s age, although a muscle sample obtained in a 11 months-old patient already showed morphological changes [6]. The important message is that a muscle biopsy with minimal changes does not preclude the diagnosis of MYMK-related myopathy.
In conclusion, MYMK-related myopathy should be suspected in patients with congenital hypotonia, motor and growth delay and a typical facial appearance due to the combination of weakness and dysmorphism. The morphological findings are not specific and muscle biopsy may show very mild involvement. Clinical course remains fairly stable, but the identification of new well characterized genetic cases will help to delineate the complete phenotype.
CONFLICT OF INTEREST
The authors report no disclosures.
Footnotes
ACKNOWLEDGMENTS
The authors thank the patient and his family for giving them permission to share his data.