
Abstract
A 38-year-old pregnant woman presented at 30 weeks gestation in respiratory distress with pre-eclampsia. This was on the background of slowly progressive dyspnoea over six years, with generalised weakness and previous surgery for ptosis and prognathia. After successful caesarean delivery at 31 weeks, the patient was found to have a homozygous likely pathogenic variant in the MYOD1 gene. This case presents a milder phenotype for MYOD1 congenital myopathy, usually associated with diaphragmatic defects, respiratory insufficiency and dysmorphic facies. It also highlights the difficulties of managing an undiagnosed patient through pregnancy.
INTRODUCTION
The Myogenic Differentiation 1 (MYOD1) gene encodes the skeletal muscle myogenic determination factor, MyoD- essential for the differentiation and repair of muscle [1]. Previously described cases with a homozygous mutation in this gene have had a severe phenotype of a congenital myopathy with diaphragmatic defects, respiratory insufficiency and dysmorphic facies (MYODRIF), with the first three described cases suffering neonatal death [2]. The clinical phenotype includes triangular facies, ptosis and prognathia, respiratory muscle weakness with elevated diaphragm domes, generalised muscle weakness, clinodactyly and renal anomalies [2–4].
Here, we report a patient with a milder phenotype, presenting during pregnancy and the successful management of her delivery.
CASE PRESENTATION
A 38-year-old pregnant woman presented at 30 weeks gestation with respiratory distress and pre-eclampsia. She had a history of slowly progressive dyspnoea over the previous six years, which was exacerbated by pregnancy. She had congenital ptosis with bilateral blepharoplasties at the age of 8 and had bilateral maxillary osteotomies for prognathism in her early 20s. She was born pre-term in Afghanistan to consanguineous parents, with a low birth weight of 1900 g, but did not require neonatal intensive care, and had an otherwise normal development. She has three siblings who are otherwise well. As a child, she was able to participate in school sports with no limitations, and had no academic difficulties, later completing a university degree.
She initially sought medical attention four years prior, for investigation of exertional dyspnoea, and was found to have chronic hypercapnic respiratory failure. At that time, she complained of shortness of breath when climbing stairs or walking long distances, without any diurnal variation, and was unable to lie flat. She was also experiencing mild dysphagia with infrequent coughing/choking episodes after eating. She denied any significant muscle weakness, diplopia or speech difficulties, and her ptosis was static. Unfortunately, the patient was lost to follow-up and moved interstate prior to any further investigations being performed.
After conceiving to her non-consanguineous husband, the patient re-engaged with medical services, with uneventful first and second trimesters, able to walk two kilometres before feeling short of breath. At 30 weeks gestation, she developed hypertension, visual disturbances and shortness of breath, requiring admission to hospital for severe pre-eclampsia.
On admission, she had a respiratory rate of 30 when sitting, oxygen saturations of 97%, blood pressure of 170/100 mm Hg and heart rate of 97 in sinus rhythm. She had a slight build, of short stature, with a prominent thoracic kyphoscoliosis and triangular facies (see Fig. 1). Her cranial nerve examination revealed bilateral ptosis without ophthalmoplegia or fatigability. There was only mild weakness of eye closure, thought to be secondary to previous blepharoplasties, with no other facial weakness or dysarthria. There was mild weakness of neck flexion. Her upper limbs were slim, with relatively small hands (see figure), reduced muscle bulk proximally and distally, and scapular instability without overt winging. Muscle tone was normal. Shoulder abduction and internal/external rotation were weak, with medical research council (MRC) power grading of 4/5, as was wrist flexion and finger extension. In the lower limbs there was wasting of the medial head of gastrocnemius and weakness around the hip girdle and ankle plantarflexion (MRC 4/5). She had brisk reflexes throughout with spread, thought to be secondary to pre-eclampsia, and flexor plantar responses. She used a Gower’s manoeuvre when getting up from a crouch. There was no dysmetria or ataxia and sensory examination was normal. Chest auscultation was clear and there was no significant respiratory muscle use. She had a gravid uterus with a cephalic foetus.
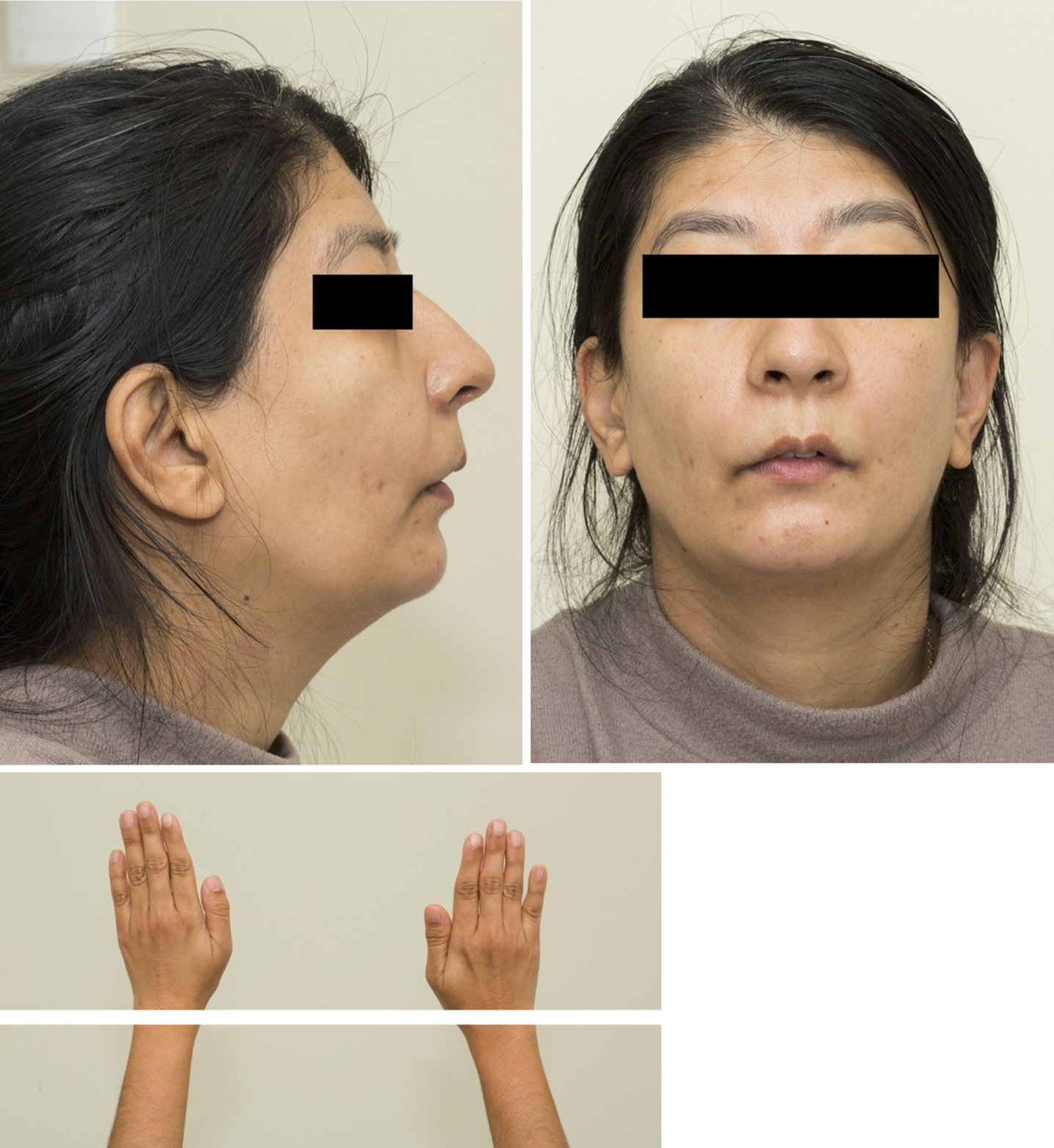
Photographs of the patient (face and hands). Published with patient’s consent.
The study was conducted in accordance with the Declaration of Helsinki. The patients provided consent for genetic analysis and data publication.
RESULTS
Spirometry showed a Forced Expiratory Volume (FEV1) of 0.77 L (35% predicted) and a forced vital capacity (FVC) of 0.85 L (27% predicted). Her electromyogram (EMG) showed normal nerve conduction studies, with no decrement on repetitive stimulation, and normal motor units and recruitment on needle examination of deltoid, triceps and biceps brachii muscles. Transthoracic echocardiogram showed normal ventricular size and function and no valvular abnormalities and a 12-lead electrocardiogram was normal. Plain film x-ray of the chest showed bilateral elevation of the diaphragm and thoracic scoliosis.
Due to worsening biochemistry (alanine aminotransferase 636 U/L, aspartate transaminase 328 U/L, lactate dehydrogenase 315 U/L, but normal platelet count, renal function and haptoglobin levels), the decision was made for elective lower uterine segment caesarean section (ELUSCS) under spinal anaesthesia with bi-level positive airway pressure (BiPAP) at 31.1 weeks gestation. The differential diagnoses considered at this point were, congenital myasthenia because of the bilateral ptosis and proximally predominant weakness, a recessive congenital myopathy, or a mitochondrial myopathy, given short stature and worsening of her weakness during the metabolic stress of pregnancy.
The patient had an uncomplicated delivery, with the baby weighing 1580 g with an Apgar score of 7, requiring admission to the neonatal intensive care unit. A concurrent muscle biopsy during delivery was offered, but the patient refused. Post-partum FVC improved to 1.15 L (35%) but reduced to 0.54 L when supine, indicating significant diaphragmatic muscle weakness and she was commenced on nocturnal BiPAP. Her creatine kinase at the time was 73 U/L. A trial of pyridostigmine was performed post-partum for possible congenital myasthenia, with FVCs measured before and after therapy showing a decrement from 1.25 L to 1.05 L and the patient reported feeling subjectively more dyspnoeic, although there was no change in clinical observations. Eight weeks later, genetic testing using a customised neuromuscular gene panel by massively parallel sequencing (MPS) revealed an apparent homozygous likely pathogenic variant in the MYOD1 gene (c.697 G > T, p.(Glu233*)). At six-month follow-up, her limb strength had improved, but with ongoing requirement for nocturnal BiPAP.
DISCUSSION
Our case expands the MYOD1 myopathy phenotypic spectrum. Affected individuals usually present at birth with hypotonia and respiratory insufficiency associated with high diaphragmatic domes on imaging [2–4]. Additional features include poor overall growth, pectus excavatum, dysmorphic facies, and renal anomalies. Others have more severe hypotonia associated with distal arthrogryposis and lung hypoplasia, resulting in neonatal death (see Table 1) [2–4]. At the very least, delayed motor development with decreased endurance would be expected, but our patient described participating in school sport with no limitation. Our patient reached adulthood prior to requiring any respiratory support and only developed clinically evident proximal weakness during pregnancy. A muscle biopsy was not performed, preventing histopathological confirmation of myopathy.
Comparison of clinical features in documented cases of MYOD1 myopathy
Citations 2 - 4: see references list. Abbreviations: BiPAP = Bi-level positive airway pressure. ND = not done.
The homozygous likely pathogenic nonsense variant of our patient is absent in the gnomAD database [5]. It is situated close to the end of the penultimate exon, likely producing a truncated MYOD1 protein with some residual function, as compared to complete protein absence due to the homozygous nonsense variant in exon 1 seen in the lethal neonatal cases of Watson et al. [2]. This is the same variant as described by Lopes et al., which also had a non-lethal phenotype [4]. Parental DNA was not available for testing. The apparent homozygosity raises two possibilities, either of which would be consistent with the phenotype: compound heterozygosity for the variant and a large (exonic) deletion or real homozygosity. Copy number variant analysis of the MPS data did not indicate a deletion.
The other challenge of this case was the management of respiratory decompensation and pre-eclampsia in third trimester pregnancy, in a patient without, at that time, a genetic diagnosis. Assessment of respiratory and cardiac function, as well as muscle weakness, and its implications for labour and anaesthesia (general or spinal) were all considerations when making management decisions. The consanguinity of the proband’s parents, and unaffected siblings, made a recessive condition more likely and therefore more likely the foetus would be unaffected. Initially a muscle biopsy was planned prior to delivery, in the hopes of guiding management, but the patient refused this. A multi-disciplinary team of obstetrics, anaesthesia, respiratory and neurology concluded that ELUSCS performed at 30 degrees head elevation with carefully titrated spinal anaesthesia and BiPAP was the safest method of delivery. The patient was counselled on the maternal risks of future pregnancies after the successful delivery of her baby.
This case broadens the MYODRIF phenotype, and highlights the issues of decompensation of undiagnosed congenital myopathies in pregnancy.
ACKNOWLEDGMENTS AND CONFLICT OF INTEREST
The authors have no conflict of interest to report.