
Abstract
Background:
The ryanodine receptor 1 (RyR1) is a major skeletal muscle calcium release channel located in the sarcoplasmic reticulum and involved in excitation-contraction coupling. Variants in the gene encoding RyR1 have been linked to a range of neuromuscular disorders including myopathies and malignant hyperthermia (MH).
Objective:
We have identified three RYR1 variants (c.1983 G>A, p.Trp661*; c.7025A>G, p.Asn2342Ser and c.2447 C>T, p.Pro816Leu) in a family with a suspected myopathy and associated malignant hyperthermia susceptibility. We used calcium release assays to functionally characterise these variants in a recombinant system.
Methods:
Site-directed mutagenesis was used to introduce each variant separately into the human RYR1 cDNA. HEK293-T cells were transfected with the recombinant constructs and calcium release assays were carried out using 4-chloro-m-cresol (4-CmC) as the RyR1 agonist to investigate the functional consequences of each variant.
Results:
RYR1 c.1983 G>A, p.Trp661* resulted in a non-functional channel, c.7025A>G, p.Asn2342Ser in a hypersensitive channel and c.2447 C>T, p.Pro816Leu in a hypersensitive channel at higher concentrations of 4-CmC.
Conclusions:
The p.Trp661* RYR1 variant should be considered as a risk factor for myopathies. The p.Asn2342Ser RYR1 variant, when expressed as a compound heterozygote with a nonsense mutation on the second allele, is likely to result in MH-susceptibility. The role of the p.Pro816Leu variant in MH remains unclear.
INTRODUCTION
Variants in the ryanodine receptor 1 gene (RYR1) have been linked to a range of congenital muscle disorders that can be inherited in either a dominant or recessive fashion. These include central core disease (CCD), multimini-core disease (MmD) [1], centronuclear myopathy (CNM) [2], King Denborough syndrome [3] and congenital fibre type disproportion [4]. The clinical symptoms for these myopathies range from a very mild phenotype with almost no muscle weakness to very severe muscle weakness with patients being unable to walk [5]. The symptoms can also differ between family members because of variable penetrance. RyR1-related myopathies are diagnosed by muscle biopsy; CCD is diagnosed by the lack of mitochondrial and oxidative enzyme activity in the central region (core) of skeletal muscle fibres [6], while other RyR1-related myopathies show increased internal and/or central nuclei, fibre-type disproportion [4] and oxidative abnormalities [7]. In recessively inherited RyR1-related myopathies compound heterozygosity has been observed: these include missense mutations together with frameshift, nonsense or splice site variants [5] with concomitant reduction in the amount of RyR1 protein [8, 9].
Patients with RyR1-related myopathies are generally considered to be susceptible to malignant hyperthermia (MH). MH is a rare autosomal dominant pharmacogenetic disorder [Online Mendelian Inheritance in Man (OMIM #145600)] that affects calcium regulation in skeletal muscle. Exposure of susceptible individuals to volatile halogenated anaesthetics or depolarising muscle relaxants can lead to tachycardia, hyperthermia, muscle rigidity and hypermetabolism and if not treated can lead to death. In addition to being linked to variants in the RYR1 gene, MH has also been linked to variants in the genes encoding the α1 S subunit of the skeletal muscle dihydropyridine receptor (CACNA1S) [10–12] and STAC3 [13]. The ryanodine receptor 1 (RyR1) is a homotetrameric calcium release channel that is located in the sarcoplasmic reticulum (SR) of skeletal muscle and plays a crucial role in excitation-contraction (EC) coupling. EC coupling is a highly regulated process with calcium release from the sarcoplasmic reticulum to the cytosol initiated by the physical interaction between RyR1 and the dihydropyridine receptor (DHPR, L-type voltage-dependent calcium channel) which is located in the transverse tubule (t-tubule) to initiate muscle contraction. STAC3 is thought to be important for the stable interaction between RyR1 and the DHPR [14, 15]. When MH-susceptible patients are exposed to triggering agents, an increase of cytoplasmic calcium in skeletal muscle fibres occurs, leading to prolonged muscle contraction and a hypermetabolic state. Therefore diagnosis of MH-susceptibility prior to general anaesthesia is essential so that triggering agents can be avoided. The in vitro contracture test (IVCT) and the caffeine halothane contracture test (CHCT) are the gold standards for the diagnosis of MH-susceptibility [16, 17]. Both tests require invasive surgery to obtain muscle biopsies from patients. The alternative is to use a DNA test where a familial causative variant has been identified. These tests are currently limited in number as to date, while over 300 variants in RYR1 have been associated with MH, only 48 have been classed as pathogenic and thus can be used diagnostically (www.emhg.org [18]).
One of the criteria to class a variant in RYR1 as pathogenic for MH is to demonstrate that the variant will result in a gain-of-function or hyperactive channel consistent with the MH phenotype. HEK293-T cells do not express a functional form of RyR1 [19], therefore they have been used widely as a system to determine the impact of RYR1 variants on calcium release [20–22].
We have used targeted exon capture and ion semiconductor DNA sequencing (Ion torrentTM) to identify three RYR1 variants in one New Zealand family with a suspected myopathy that appeared to be associated with MH-susceptibility. This was followed by functional characterisation of each of these variants in the HEK293-T system.
PATIENTS AND METHODS
Ethical approval
Human blood and muscle tissue were obtained from patients with informed consent and approval by the Central Region (Wellington, New Zealand) human ethics committees (MWH0010051 or MWH/03/04/018). The study has been approved by the Massey University Genetic Technology committee (GMO 05/MU/01 and GMO 00/MU/60) acting as an Institutional Biological Safety Committee for the Environmental Protection authority, Wellington, New Zealand.
Clinical history
Patient AII:1 (proband, Fig. 1) underwent surgery for elective tonsillectomy at 8 years of age. She experienced severe masseter spasm (a potential indicator of MH-susceptibility [23]) after induction with suxamethonium and the remaining procedure was carried out under total intravenous anaesthesia. Creatine kinase levels on the next day were severely elevated (2934 IU). No family history of MH had been reported but a suspected myopathy in the mother (AI:1) and the proband due to elongated facial features. Both parents (AI:1 and AI:2) subsequently underwent a muscle biopsy and in vitro contracture test, as the proband was too young at the time. The histochemistry findings of the mother’s (AI:1) muscle biopsy were unremarkable except for the presence of two COX negative fibres (<2% of total fibres). No structurally abnormal mitochondria were observed on electron microscopy. The mother had one previous, unremarkable general anaesthesia with sevoflurane for 15 minutes. Neither the father nor the sister of the proband (2 years younger than proband) have had general anaesthesia. The sister (AII:2) does not have elongated facial features.
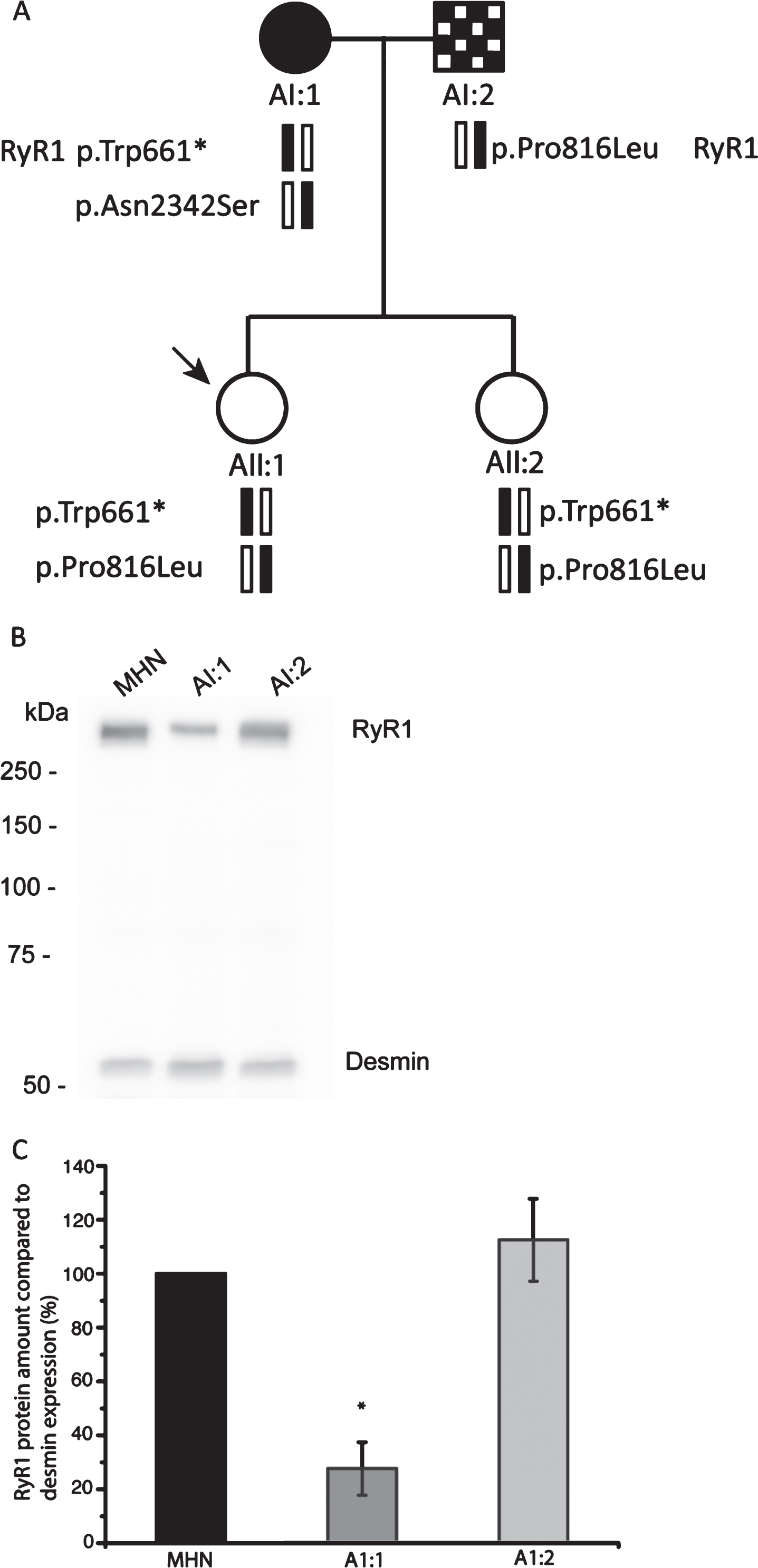
Segregation analysis of family A and expression of RyR1 protein in patient skeletal muscle.
In vitro contracture testing
Muscle biopsies and in vitro contracture tests were performed at Palmerston North hospital according to the European Malignant Hyperthermia Group (EMHG) guidelines [16] and local laboratory control data [24]. A MH-susceptible (MHShc) diagnosis is made if the response to both halothane (2%) and caffeine (2 mM) is above the threshold of 0.4 g and 0.2 g respectively. If the response is below the threshold for both agents the patients are diagnosed as MH-negative (MHN). If the response for only one agent is above the threshold, the patients are diagnosed as MHSh or MHSc for halothane and caffeine respectively. MHShc, MHSh and MHSc patients are all treated as susceptible to MH in a clinical situation.
Capture and sequencing of genomic DNA/data analysis
Capture and DNA sequencing was performed at Lotterywest State Biomedical Facility Genomics, University of Western Australia, Perth for samples AI:1, AI:2 and AII:1. Genomic DNA (1μg) was used for hybridization to a custom TargetSeq kit (Life Technologies, 336 neuromuscular disease genes Supplementary Table 2). The probes target all exons, including 5’ and 3’ UTRs (untranslated region) spanning approximately 3.2 Mb. The DNA was initially sheared to ∼180bp fragments using a Covaris S2 Ultrasonicator. Barcoded sequencing adaptors were attached using the NEXT Ultra kit (New England Biolabs). The barcoded samples were pooled and size-selected on a 2% agarose gel. The samples were then attached to Ion Torrent ISPs (beads) using an Ion Proton 200bp OneTouch2 templating kit (Life Technologies), enriched and sequenced on a single Ion P1 sequencing chip for 500 flows. Mapping and variant calling was performed using TorrentSuite 4.0, with high-stringency variant-calling parameters which typically yield 1200-1400 variants per sample. The variants were then annotated using Annovar [25]. RYR1 exon 91 could not be captured efficiently because of the high GC content and was subsequently analysed by Sanger sequencing. Classification of variants is according to GenBank accession numbers NM_000540.2 and NP_000531.2 for RYR1 and NM000069.2 and NP_000060.2 for CACNA1S for cDNA and protein, respectively. Sample AII:2 was screened for all variants identified in samples AI:1, AI:2 and AII:1 in RYR1 and CACNA1S. As STAC3 was not included in the neuromuscular gene panel, all exons of this gene were sequenced from genomic DNA from all family members (primer sequences available on request).
Cloning of mutant RYR1 constructs
The full length human RYR1 cDNA had been previously cloned into pcDNA3.1(+) [26]. Site-directed mutagenesis was used to introduce variants into human RYR1 cDNA subclones using the QuikChange™ method (Stratagene) according to the manufacturer’s instructions. Primer sequences can be found in Supplementary Table 1. Sanger sequencing was used (Massey Genome Service, Palmerston North, New Zealand) to confirm the variants in the recombinant constructs as well as the absence of PCR-induced errors. Subsequent subcloning steps were used to create full length RYR1 cDNA constructs in pcDNA3.1(+).
Transient expression of RyR1 variants in HEK293-T cells
HEK293-T cells were maintained in high glucose Dulbecco’s modified Eagle’s medium (Sigma) containing 10% foetal calf serum (Sigma) and 0.5% penicillin/streptomycin (Life Technologies). Fugene HD (Promega) was used for transfection of HEK293-T cells with pcDNA3.1(+) containing wildtype or mutant RYR1 cDNA at a ratio of 8 : 2 for Fugene HD (μL) to plasmid DNA (μg). Cells were harvested 72 hours after transfection for western blotting or used directly for calcium release assays.
DNA and RNA extraction, Reverse Transcriptase PCR, Sanger sequencing
Genomic DNA from blood was extracted using the Wizard™ Genomic DNA Purification kit (Promega) according to the manufacturer’s instructions. Total RNA was extracted from 100 mg frozen skeletal muscle tissue using TRizol® Reagent (Invitrogen Life Technologies) according to the manufacturer’s instructions. Total RNA was first treated with TURBO Dnase (Ambion), then 1.5μg RNA was used to synthesize first strand cDNA using the Transcriptor First-Strand cDNA synthesis kit (Roche) according to the manufacturer’s instructions. Specific regions in gDNA or cDNA were amplified by polymerase chain reaction (PCR, primer sequences can be found in Supplementary Table 1). PCR products were purified using the Wizard® SV gel and PCR clean-up system (Promega) and subsequently sequenced using BigDye™ Terminator V3.1 on an ABI 3730 (Applied Biosystems, Massey Genome Service, Palmerston North) DNA analyser.
High resolution amplicon melting analysis
Over 140 MHN gDNA samples were screened for the presence of the RyR1 p.Pro816Leu, p.Asn2342Ser and p.Trp661* variants using high resolution amplicon melting analysis (HRM) as described previously [27]. Real-time PCR and HRM analysis were performed on the Roche LightCycler 480 system using the LightCycler® 480 High Resolution Melting Master mix (Roche). Primer sequences can be found in Supplementary Table 1. PCR conditions are described in Supplementary Material.
Functional calcium release assays
Calcium release assays using HEK293-T cells were performed as previously described [21]. HEK293-T cells were washed once with 1x BSS buffer (140 mM NaCl, 2.8 mM KCl, 1 mM MgCl2, 10 mM HEPES, 2 mM CaCl2, 10 mM glucose, pH 7.3), loaded with fura-2/AM (4μM final concentration, Invitrogen) and 0.05% pluronic F-127 (Sigma) in 1x BSS buffer for 1 hour at 37°C in the dark. Cells were washed once in 1x BSS buffer, then in calcium-free 1x BSS buffer containing 2 mM EGTA. Ca2 + release assays were carried out using a fluorescence microscope (Olympus IX81, Tokyo, Japan) after addition of incremental concentrations of 4-chloro-m-cresol (4-CmC, Sigma). Activation of RyR1 was measured by the change in fluorescence emission ratio at 510 nm when excited at 340 nm and 380 nm. Results were calculated as mean values (±SEM) of n (n = 6-12) results for each 4-CmC concentration and EC50 (50% effective concentration) values were calculated from sigmoidal curves (fitted using Origin software, v. 8.5.1, OriginLab Corporation) from individual replicates. Statistical analyses were performed using two-way ANOVA or the Student t test for paired samples. Bonferroni correction for multiple tests was carried out to confirm statistical significance.
Western blotting
Total protein from HEK293-T cells transiently expressing RyR1 was extracted in cell lysis buffer (0.1 M Tris pH 7.8, 0.5% Triton X-100) freshly supplemented with 1x cOmplete™ Mini protease inhibitor (Roche). Frozen muscle biopsy tissue (200 mg) was homogenised in cell lysis buffer (0.1 M Tris pH 7.8, 0.5% Triton X-100) supplemented with 1x cOmpleteTM Mini protease inhibitor (Roche). Protein assay dye reagent (Bio-Rad) based on the Bradford assay [28] was used to determine the protein concentration of the samples. Western blotting was performed using standard protocols [29]. Briefly, solubilized protein (∼20μg for patient muscle protein, 200μg for HEK293-T cells) was fractionated on a 7.5% SDS-PAGE gel and transferred overnight onto a polyvinylidene difluoride (PVDF) membrane. The monoclonal RyR1 34 C antibody (Sigma) or polyclonal H-21 antibody (Santa Cruz Biotechnology) were used to detect RyR1 protein (full length or N-terminal region, respectively); the monoclonal T9026 (Sigma) antibody was used to detect α-tubulin (for HEK293-T cells), the polyclonal PA5-16705 (Thermo Fisher) antibody was used to detect desmin (for skeletal muscle cells) followed by horse radish peroxidase conjugated anti-mouse (A-9044, Sigma, for 34 C and tubulin antibodies), anti-goat (A5420, Sigma, for H-21) or anti-rabbit (W4011, Promega, for desmin) secondary IgG antibodies.
RESULTS
IVCT data, targeted DNA sequencing and variant screening
Both parents of the proband who experienced a suspected MH reaction underwent a muscle biopsy and subsequent IVCT (Table 1, Fig. 1A). The mother of the proband (AI:1) was diagnosed as MHShc, whereas the father (AI:2) was diagnosed as MHSh.
In vitro contracture test (IVCT) data of family A. Maximal contractures in IVCT at the threshold concentrations of caffeine (2 mmol litre-1) and halothane (2%)
These findings prompted targeted exon DNA capture (336 neuromuscular disease genes) and next generation sequencing (NGS) (Supplementary Table 2) for the parents and proband. Average target depth (reads) for AI:1, AI:2 and AII:1 were 130, 95 and 89 respectively. Percentage of target bases with at least 20x read depth for samples AI:1, AI:2 and AII:1 were 89%, 86% and 86% respectively. Coverage for CACNA1S and RYR1 were 100% with the exception of RYR1 exon 91 which was sequenced by Sanger sequencing. STAC3 was not included in the neuromuscular gene panel and was sequenced using Sanger sequencing. Numerous variants were identified in the samples (Table 2) including several in RYR1 (Table 3). The mother (AI:1) of the proband carried the RYR1 variants c.1983 G>A, p.Trp661*[5], c.7025A>G p.Asn2342Ser [30] and c.11266 C>G p.Gln3756Glu [31]. The father (AI:2) of the proband carried the RYR1 variants c.2447 C>T, p.Pro816Leu and c.10747 G>C, p.Glu3583Gln [31]. The proband (AII:1) inherited the c.2447 C>T, p.Pro816Leu from her father and the c.1983 G>A, p.Trp661* and c.11266 C>G p.Gln3756Glu variants from her mother. Therefore the p.Trp661* and c.11266 C>G p.Gln3756Glu variants are on the same allele and so the latter would not be expressed. All variants in RYR1 were confirmed by Sanger sequencing of genomic DNA. The sister of the proband (AII:2) carried the same RYR1 variants as the proband AII:1 (Table 3). Two common variants in CACNA1S were identified in AI:2 and AII:1 (Table 3). The c.1983 G>A, p.Trp661*, c.7025A>G p.Asn2342Ser and c.2447 C>T, p.Pro816Leu variants were not detected in any of the 140 MHN control samples screened by HRM, as would be expected for rare variants.
Variants identified in family A by DNA capture and NGS in 336 neuromuscular disease genes
Nonsynonymous variants identified in RYR1 and CACNA1S in family A
All variants in RYR1 are according to GenBank accession NM_000540.2 and NP_000531.2 for cDNA and protein respectively. All variants in CACNA1S are according to GenBank accession NM_000069.2 and NP_000060.2 for cDNA and protein respectively. aMinor Allele frequency from gnomAD https://gnomad.broadinstitute.org/.
Expression of the RYR1 variants in patient cells
Heterozygous endogenous expression of the c.2447 C>T, p.Pro816Leu variant was confirmed in muscle tissue by sequencing cDNA prepared from RNA isolated from muscle tissue of AI:2 (Supplementary Figure 1). The c.7025A>G, p.Asn2342Ser variant was expressed monoallelically in muscle tissue in AI:1 (Supplementary Figure 1) and the c.1983 G>A, p.Trp661* variant was expressed at very low levels only. To check for haploinsufficiency in the presence of the premature stop codon (c.1983 G>A, p.Trp661*), total protein was isolated from muscle biopsy tissue of AI:1, AI:2 and one non-related MHN control and RyR1 levels detected using semi-quantitative western blotting, with desmin [32] as a loading control. The relative amount of RyR1 protein in AI:1 was 27.6±9.9% (n = 4) and 112.4±15.4% in AI:2 (n = 4) compared to an MHN control (Fig. 1B and C).
Functional analysis of variants
Recombinant RyR1 expression and calcium release assays
Western blotting confirmed expression of all the mutant and WT RyR1 proteins (Fig. 2) in transiently expressing HEK293-T cells, using tubulin as a loading control, p.Thr2787Ser was included as a benign variant for an additional control (Supplementary Table 3) [33]. No RyR1 protein was detected in the mock control (cells transfected with empty pcDNA3.1 vector). Transiently expressing HEK293-T cells loaded with fura-2/AM were exposed to increasing concentrations of 4-CmC and Ca2 + measured using a fluorescence microscope. The data were normalized to Ca2 + released at 1000μM 4-CmC. Figure 3 shows the sigmoidal fitted curves for the three variants (p.Trp661*, p.Pro816Leu and p.Asn2342Ser) and three controls (wild-type, p.Arg533Cys and p.Thr2787Ser). In the presence of the p.Trp661* variant, no Ca2 + was released at any concentration of 4-CmC. The concentration-response curve for the p.Asn2342Ser variant (and the p.Arg533Cys positive control) showed a significant shift to the left compared to the wild-type control. Ca2 + release for the p.Pro816Leu variant compared to wild-type increased only at concentrations higher than 400μM 4-CmC. The p.Thr2787Ser variant showed no significant difference in Ca2 release when compared to wild-type RyR1 as would be expected for a benign variant. EC50 values are shown in Table 4.
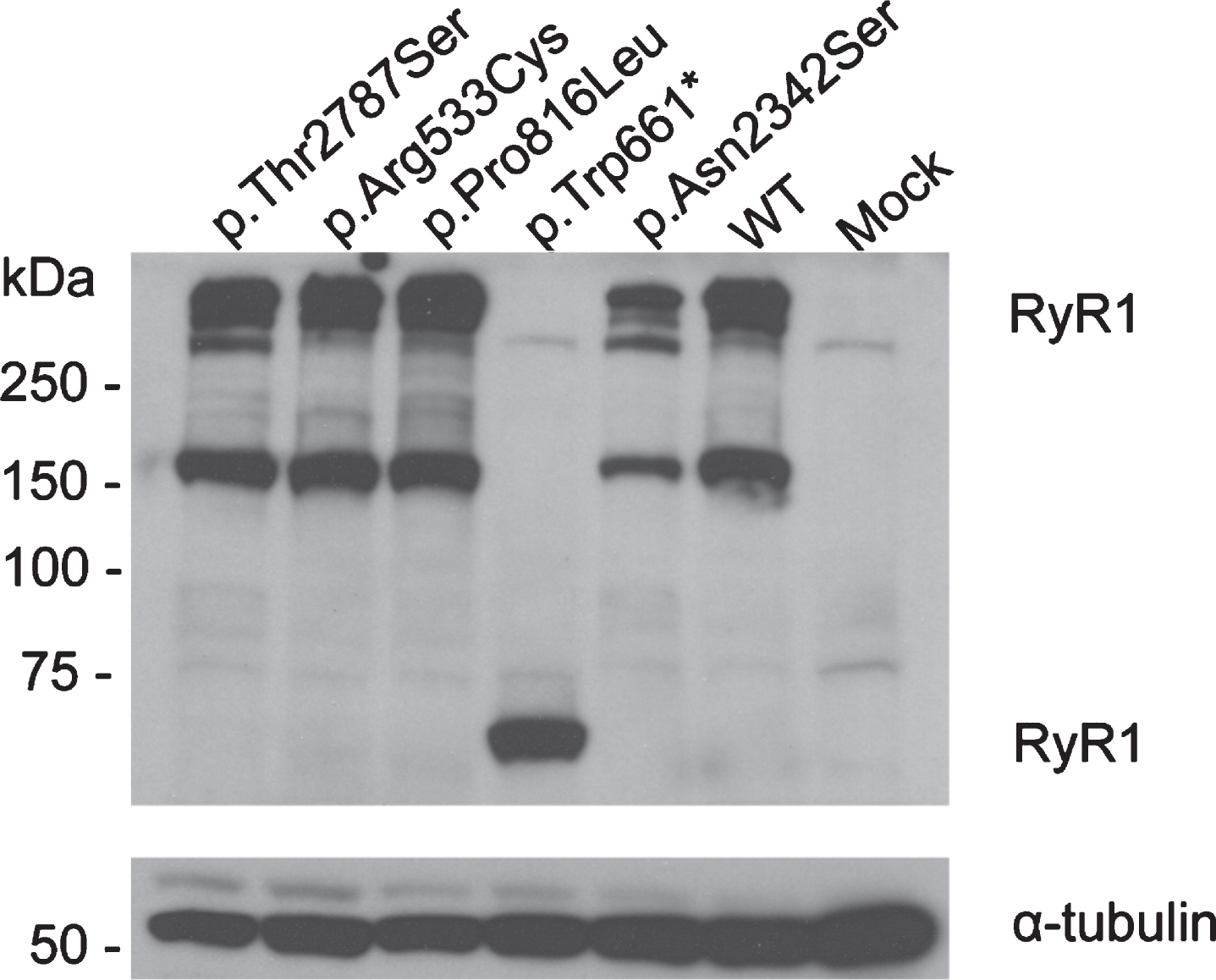
Expression of RyR1 protein in HEK293-T cells. Western blot showing expression of RyR1 protein in transiently transfected HEK293-T cells. Total protein extract was loaded onto a 7.5% polyacrylamide gel and blotted onto PVDF membrane. RyR1 and α-tubulin were detected using 34 C and anti-α-tubulin antibodies. Variants are indicated by their amino acid change, WT: wild-type.
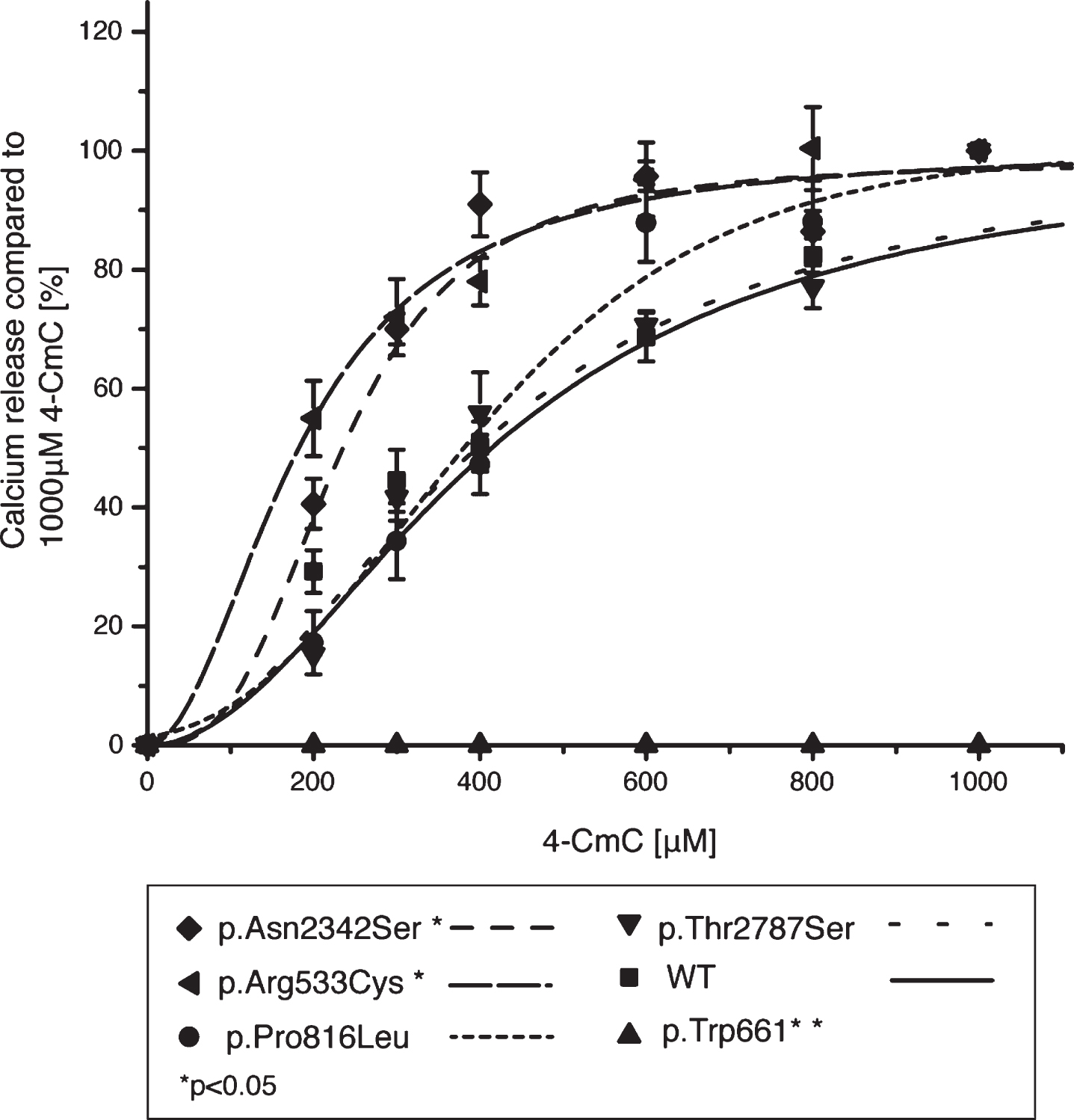
Normalized 4-chloro-m-cresol (4-CmC) concentration-response curves for transiently expressing HEK293-T cells. Ca2 + release upon stimulation with 4-CmC is expressed as the percentage of maximal Ca2 + released by 1000μM 4-CmC. Concentration-response curves were generated using Origin software, results are shown as mean±SEM (n = 6-12). The cells carrying the p.Asn2342Ser variant show increased sensitivity to 4-CmC compared with wild-type (WT) control cells.
EC50 values for 4-CmC induced calcium release in transiently transfected HEK293-T cells
*Statistical significance, p < 0.01.
DISCUSSION
So far over 300 variants in RYR1 have been associated with MH-susceptibility. The advent of new sequencing technologies has resulted in the identification of new variants not only in RYR1 but also in other genes. Although segregation of the variants with the disease in families is an important factor, functional studies ex vivo and/or preferably in vitro still remain an essential requirement to demonstrate that a variant is pathogenic for MH-susceptibility. The aim of this study was to identify the cause of MH and a potentially associated myopathy in a New Zealand family. Using a targeted gene panel and next generation sequencing, we identified three RYR1 variants in the proband which she had inherited from her parents. The RyR1 p.Gln3583Glu and p.Glu3756Gln variants have relatively high allele frequencies of 0.0147 and 0.0331, respectively in the gnomAD browser (https://gnomad.broadinstitute.org/). These two variants were not investigated further as they do not segregate with MH-susceptibility in previous studies [31] although a modifier effect cannot be ruled out.
The p.Trp661* variant in the proband, inherited from her mother, had been described previously in a patient in combination with a RyR1 p.Met2423Lys variant [5]. The patient in this study had generalized weakness, marked facial weakness and multiple cores [34]. The proband and her sister in our study carried the p.Trp661* variant in combination with the p.Pro816Leu variant. The proband’s mother carried the p.Trp661* variant in combination with the p.Asn2342Ser variant. The p.Trp661* variant shows complete lack of activity using functional analysis in HEK293-T cells, and a very low level of endogenous expression (Supplementary Figure 1). Therefore it is likely that the RYR1 alleles being expressed in vivo are the p.Pro816Leu variant (proband) and p.Asn2342Ser variant (mother). At least in the case of the mother, haploinsufficiency was observed in terms of RyR1 expression. Although we could not measure RyR1 protein levels in the proband as muscle tissue was not available, it is possible that a similar haploinsufficiency exists with expression of the p.Pro816Leu variant. The suspected myopathy in the proband and her mother may be due to lower levels of RyR1 protein. The MH-susceptibility observed in the mother is likely due to monoallelic expression of the p.Asn2342Ser variant, as this showed hypersensitivity to agonist in our functional assays, consistent with an MH-susceptible phenotype. The proband and her sister both carry the p.Pro816Leu variant but the sister has no recognizable myopathy. It is possible that phenotypic symptoms of a myopathy are too mild to be detected, or have not yet arisen in the sister. The proband is now 16 years old, her sister (AII:2) 14 years old. Alternatively, the suspected myopathy in the proband and her mother could be caused by variants in genes other than RYR1, although no obvious candidates were identified in the neuromuscular panel genes that were screened.
The p.Asn2342Ser variant has been described before in association with MH [30]. Functional analysis in B-lymphoblastoid cells from a patient (by measuring metabolic proton release after stimulation with the RYR1 agonist 4-CmC) suggested pathogenicity of the variant [35]. This method of assessing variants potentially associated with MH-susceptibility however, has not yet been accepted by the EMHG. It is possible that the genetic background of the patient-derived lymphocytes could have influenced these results. The p.Asn2342Ser variant was also found in the three MHN sons of a father with MH-susceptibility in a different study, suggesting the variant may not be pathogenic [36]. In a study of 870 exomes from unselected volunteers the p.Asn2342Ser heterozygous variant was found twice and classed as likely not pathogenic based on a survey of the relevant literature [33]. The variant has also been found in 8 families in the UK, but segregates with MH in only one family [37]. This variant has a minor allele frequency of 0.00102 in the gnomAD browser (accessed October 2019), which is just over the predicted incidence of 0.1% suggested for a variant pathogenic for MH-susceptibility [31]. In silico pathenogenicity prediction suggests that this variant could have an impact on protein function (Supplementary Table 3). It is located in one of three so called “hot-spot” regions in RYR1 and located close to the diagnostic p.Arg2336His variant. This region of the protein has been predicted to form functional contacts with the N-terminal region, but only the backbone, rather than individual residues were able to be traced in the cryo-EM structure of rabbit RyR1 [38]. At present, any definite role for the p.Asn2342Ser variant must remain speculative. Nevertheless, our functional studies in HEK293-T cells show that RyR1 carrying this variant releases calcium at lower concentrations of 4-CmC compared to wild-type RyR1, suggesting it could have a significant role in channel function if expressed monoallelicly.
The p.Pro816Leu variant has been identified in one MH-family in the United Kingdom, but does not segregate with MH in this family [37] and has a minor allele frequency of 0.0000212 in the gnomAD browser. We show that RyR1 containing the p.Pro816Leu variant released more calcium only at higher concentrations of agonist compared to wild-type RyR1. The p.Pro816Leu is located in the SPRY1 domain of RyR1 and is highly conserved (Supplementary Figure 2) between species and RyR isoforms. In silico pathogenicity prediction using multiple models suggests that this variant could be disease causing (Supplementary Table 3). The SPRY1 domain has been predicted to be involved in FKBP12 binding [39] which has been predicted to stabilize the closed state of the RyR1 channel. The interaction between FKBP12 and RyR1 may be compromised by the p.Pro816Leu amino acid change, but further experiments would be required to address this possibility and its involvement in MH-susceptibility cannot be ruled out based on the collective evidence presented here. The father of the proband was MHSh (only responding to halothane in the contracture test). A recent study by Figueroa et al. [40] suggested that the underlying cause of MH in individuals responding only to halothane, but not caffeine in contracture testing, involves a different mechanism compared to individuals responding to both halothane and caffeine.
In the HEK293-T cell system, individual variants in a gene can be functionally compared to each other in an identical genetic background. This is an advantage compared to the patient derived lymphocyte or myoblast systems where the patient’s genetic background could influence results. The disadvantage of the HEK293-T system is however that the variant is expressed in a homozygous manner, while the majority of MH-susceptible patients are heterozygous for the variants. As cDNA sequencing from muscle tissue from the mother of the proband suggested expression of only the p.Asn2342Ser variant, it is also likely that the p.Pro816Leu variant is also expressed in this manner in the proband.
CONCLUSIONS
The p.Asn2342Ser RYR1 variant, when expressed as a compound heterozygote with a nonsense mutation on the second allele, is likely to result in MH-susceptibility and therefore should be considered pathogenic for MH, under these conditions. The role of the p.Pro816Leu variant in MH remains unclear. Expression of this variant in a system more analogous to skeletal muscle may be required to provide more definitive evidence of a role in MH. It is possible that a gene other than RYR1 may be responsible for the suspected myopathy in the proband and her mother as the only allele in RYR1 that they share carries the p.Trp661* nonsense codon or it could be due to haploinsufficiency.
ACKNOWLEDGMENTS
We would like to thank the family for their cooperation. The authors would also like to thank Keisaku Sato for construction of the full length wt RYR1 cDNA clone, Lili Rhodes for assistance in cloning the RYR1 mutant constructs and Sanger sequencing, and Shannon Ormond for help with coverage analysis. This work was supported by the Massey University Research Fund, a grant from the Palmerston North Medical Research Foundation and the Australian and New Zealand College of Anaesthetists, Melbourne, Victoria, Australia (09/013 and 15/011).
CONFLICT OF INTEREST
The authors have no conflict of interest to report.