
Abstract
BACKGROUND:
Extensive genetic screening results in the identification of thousands of rare variants that are difficult to interpret. Because of its sheer size, rare variants in the titin gene (TTN) are detected frequently in any individual. Unambiguous interpretation of molecular findings is almost impossible in many patients with myopathies or cardiomyopathies.
OBJECTIVE:
To refine the current classification framework for TTN-associated skeletal muscle disorders and standardize the interpretation of TTN variants.
METHODS:
We used the guidelines issued by the American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology (AMP) to re-analyze TTN genetic findings from our patient cohort.
RESULTS:
We identified in the classification guidelines three rules that are not applicable to titin-related skeletal muscle disorders; six rules that require disease-/gene-specific adjustments and four rules requiring quantitative thresholds for a proper use. In three cases, the rule strength need to be modified.
CONCLUSIONS:
We suggest adjustments are made to the guidelines. We provide frequency thresholds to facilitate filtering of candidate causative variants and guidance for the use and interpretation of functional data and co-segregation evidence. We expect that the variant classification framework for TTN-related skeletal muscle disorders will be further improved along with a better understanding of these diseases.
INTRODUCTION
The 364-exon TTN gene (MIM188840) encodes titin, the largest human protein [1]. It is expressed in both cardiac and skeletal muscle and the 1.5μm long molecules span from the Z disk to the M line of the sarcomere.
High throughput sequencing (HTS) strategies have enabled the genetic analysis of the whole TTN gene (2), although its large genomic size and its complex, highly repetitive structure still pose technical problems: variants in the triplicate region, for example, are often not called or miscalled by the routinely used bioinformatics tools [2].
Pathogenic variants in TTN cause different skeletal muscle diseases, both dominant and recessive, with or without cardiac involvement [2]. The clinical use of HTS technologies has further expanded the spectrum of the TTN-related skeletal muscle phenotypes, ranging from severe congenital myopathies to relatively mild adult onset muscle disorders [3–5].
Moreover, TTN truncating variants (TTNtv) have been implicated in heart muscle diseases (dilated cardiomyopathy, DCM) [6, 7]. However, in many of the studied DCM families the identified TTNtv also segregate in asymptomatic individuals [6, 7], suggesting that the penetrance of heterozygous TTNtv is not complete. The genetic and environmental factors that impact TTNtv penetrance in the setting of DCM have not yet been fully elucidated.
Because of the huge size of the gene, TTN variants, mainly missense, are identified in each HTS analysis, representing common findings. A number of missense pathogenic variants located in exons 344 and 364 have already been confirmed in TTN-related skeletal muscle disorders [5, 9], mainly because of their co-segregation with the disease in multiple unrelated families. Similarly, a small number of missense variants have been associated with a cardiomyopathy [10, 11]. Interestingly, genetic and biophysical studies have suggested the pathogenicity of a variant (p.A178D) causing a dominant, highly penetrant, cardiomyopathy with features of left ventricular noncompaction.
However, most of the TTN missense variants identified by HTS-based tests have unknown significance and their clinical evaluation in diagnostic laboratories remains challenging. As underlined by the American College of Medical Genetics and Genomics (ACMG)/Association for Molecular Pathology (AMP) recommendation [12], assessing the clinical significance of variants is a multi-step process that should take into account different resources and data. However, these guidelines should be regarded as a general framework to be adapted to each single gene/disease. In this view, TTN with its huge size, its repetitive modular structure, the wide spectrum of related disorders and the yet-to-be-determined underlying mechanisms of disease, is certainly an extraordinary case [1, 13].
The ACMG/AMP guidelines report a set of criteria supporting the benign or pathogenic classification of genetic variants [12]. In this work, we undertook a re-evaluation of these criteria for the classification of TTN variants in the context of inherited skeletal muscle disorders.
MATERIALS AND METHODS
Standard protocol approvals, registrations, and patient consents
All the patients and families provided written informed consent, according to the Declaration of Helsinki. The study was approved by Ethics Review Board of Helsinki University Hospital (number 195/13/03/00/11).
Sequencing data
In this study we analyzed the clinical and sequencing data from 2772 patients with a wide spectrum of inherited skeletal muscle diseases, including muscular dystrophies, distal myopathies, congenital myopathies and isolated hyperCKemia who underwent HTS-based tests (Supplementary Figure 1). Over 70% of them had an adult onset. The cohort of patients enrolled is composed of European non Finn (55%), Finn (40%) and other ethnicities (5%). Data from an additional 470 unaffected relatives enrolled for trio or duo analysis were also analyzed.
Four hundred eighty samples were studied by exome sequencing within the MYO-SEQ project [14]; one hundred ten samples were analyzed by exome sequencing in Ljubljana; the remaining samples were analyzed by several different targeted approaches covering TTN in seven different laboratories: Helsinki, Naples, Pisa, Brno, Milan, Bethesda and Neu-Ulm [15–19].
We did not exclude a priori patients with a confirmed diagnosis (reasonably, 20–25% of our cohort of 2722 patients), involving an alternative known muscle disease-causing gene, to avoid any possible bias related to the different variant interpretation in the different centers. In line with previously published studies, we noticed that molecular findings in TTN and in genes other than TTN were differently evaluated in the different centers.
For the aim of this study, only TTN exonic non synonymous variants or variants affecting canonical splice sites were re-evaluated.
Not all the variants were experimentally validated by Sanger sequencing. However, only variants: a) passing the GATK quality control filters (tagged as 'PASS' in GATK); b) in positions covered at least 20x; c) with a minor/wild type (wt) allele ratio higher than 0.30; and d) supported by reads in both directions have been evaluated. Moreover, considering the possible error rate associated with long insertions and deletions, all the calls were manually confirmed by inspecting the raw reads using the IGV software [20], filtering out possible false positives.
Population allele frequency calculation
The population allele frequency for potentially causative TTN variants was calculated as previously described by Whiffin and colleagues, using an online tool (https://www.cardiodb.org/allelefrequencyapp/) [21]. Their approach takes into account disease prevalence, genetic and allelic heterogeneity, inheritance mode and penetrance [21].
We calculated the population allele frequency cutoff for TTN variants responsible for a myopathy in several different scenarios, under a dominant or a recessive inheritance pattern (Table 1). Since TTN mutations can cause different skeletal muscle disorders, we included the estimated prevalence of unsolved skeletal muscle disorders (around 1/25000 individuals) [22, 23]. We considered a genetic heterogeneity of 0.1–0.3 (10–30% of unsolved myopathies due to TTN causative variants [4, 24]); an allelic heterogeneity of 0.01–0.02 (no single variant causing more than 1-2% of cases, in line with the current data [2, 26]) and a penetrance of 0.5–1 (so far, variants causing a skeletal muscle titinopathy seem to be fully penetrant [2, 26]; however, also considering that TTNtv are not fully penetrant in the context of cardiac diseases [6, 7], we used a conservative approach, considering also the possible presence of low penetrant variants causing a TTN-related skeletal muscle disorder). We calculated that TTN variants causing a recessive skeletal muscle disorder should have a MAF<9.80E10-5 (Table 1). Similarly, the higher MAF for variants causing a dominant myopathy should be 2.40× 10E-7 (for 50% penetrant variants).
General frequency of TTN candidate disease-causing variants
Re-occurring variants
n.e. = exon non included in the six TTN isoforms described in adults, p.r. = previously reported, s.p. = variants p.A30941T and p.K17004N are identified in the same two patients (therefore, probably in cis); variants p.T18595R and p.G14831E are similarly shared by other two patients (i.e. they are probably in cis), o.d. = possible other diagnosis in one of the patients, p.c. = poorly covered in exome sequencing data (gnomAD), NA = not available, * Variant p.I35947N has been seen in 2 patients. One patient also carries a TTNtv (variants not phased, see Savarese et al. 2018); Variant p.W33529R has been seen in 3 patients. Two of them also carry a TTNtv (variants not phased). The third patient (described in Savarese et al. 2018) has only missense variants on the second allele. Variant p.A10614D has been seen in 2 patients. One patient also carry a TTNtv (variants not phased).
Quasi case-control study
We compared the frequency of selected variants (re-occurring in at least two unrelated patients) in our myopathy cohort to their minor allele frequency (MAF) in gnomAD [27]. In this part of the study, we excluded variants identified in patients with a confirmed molecular diagnosis involving an alternative known muscle disease-causing gene or identified in related patients.
Only variants at genomic positions covered in gnomAD data were included in our analysis. In the absence of genome-wide data, we were not able to perform an ethnic stratification. For an unbiased evaluation, we considered the general MAF in gnomAD (release 2.1) [28] as well as the highest MAF reported for a specific population. A Fisher’s exact test was performed for comparing the frequency of mutated alleles in the patient cohort versus the gnomAD frequencies. The calculated p-values were adjusted by Bonferroni correction for multiple testing.
Sequence variants in TTN are described according to the coding DNA reference sequence (NG_011618.3 or LRG_391), covering transcript variant-IC (NM_001267550.1), commonly described as the titin metatranscript. The exon numbering is the LRG numbering, as used by the Leiden Open Variation Database (LOVD - http://www.LOVD.nl/TTN).
Variant annotation
The impact of selected missense variants on the protein structure has been evaluated using wAnnovar (http://wannovar.wglab.org/) [29] and considering the prediction of thirteen different bioinformatic tools [30–39]. We used SpliceAI to evaluate the possible splice effect of exonic variants causing missense changes, using the recommended delta score threshold of 0.5, meaning that the probability of the variant being splice-altering is over 50% [40]. We also evaluated the presence of the experimentally identified TTN variants in LOVD and ClinVar (https://www.ncbi.nlm.nih.gov/clinvar).
TITINdb (http://fraternalilab.kcl.ac.uk/TITINdb) [41] was used to map titin variants to domain structures.
RESULTS
We re-evaluated the TTN variants identified in our large multi-center cohort of 2772 patients with a skeletal muscle disorder, following the ACMG/AMP guidelines [12].
Population data
In our case cohort, 1,227 variants have a MAF lower than MAF<9.80E10-5 in the different gnomAD populations. Of these, over 80% (1,069) are missense variants and 158 are possible truncating variants (nonsense, variants in canonical splice sites and indels causing a frameshift).
Mutational hotspots
Of the 1,069 missense variants with a MAF<9.80E10-5, fifteen are located in the exon 344, associated with the hereditary myopathy with early respiratory failure (HMERF) [5, 42]. In particular, eight variants have been previously reported as being causative.
Eleven missense variants are located in exon 364, associated with a dominant tibial muscular dystrophy and with recessive non-congenital titinopathies (proximal or distal myopathy) [9, 43]. One of this (p.I35947N) is a well-known disease causing variant [13].
'Quasi case-control' study
Table 2 lists the TTN variants with a MAF<9.80E10-5 that re-occurred in at least two unrelated patients who had remained undiagnosed after preliminary HTS study. Twelve TTNtv re-occurred in unrelated patients and two – the previously reported p.K35963Nfs*9 and p.Q35879* - show a significant enrichment if compared to the frequency in gnomAD (adjusted p-value 1.52E10-4 and 6.27E10-7, respectively). Similarly, we identified 59 missense changes re-occurring in at least two independent families, including 14 previously undetected variants (not listed in gnomAD). Two missense variants (p.I20738M and p.N16170T) shows a significant enrichment with an adjusted p-value of 0.02 and 0.04, respectively. Interestingly, the previously described variant p.W33529R has been identified in 3 unrelated patients (two of them also carrying nonsense variants - phasing and segregation studies are ongoing).
Computational/predictive data and other databases
We annotated with wAnnovar the 1,069 candidate TTN missense variants (MAF<9.80E10-5), the 59 re-occurring missense variants listed in Table 2 and 59 common SNPs (MAF > 3% in gnomAD). As expected, the predictions largely vary among different programs. Interestingly, only one missense variant (p.G34002R, not listed in gnomAD), identified in a single patient, is predicted as being damaging by all the programs we used. Only two programs (Meta_SVM and Meta_LR) classified all the common SNPs as tolerated (Supplementary Table 1).
We also evaluated the possible effect of the 1,069 exonic variants, predicted as causing missense changes, on the splicing. Interestingly, SpliceAI predicts two variants to be splice-altering, including one of the re-occurring variants (c.9451A>G, p.S3151G with a delta score for donor gain = 0.8267)
Out of the 1,069 missense variants, 88 are listed in LOVD and 202 in ClinVar: only two variants (p. C31712R and p. I35947N) are tagged as pathogenic in both the databases. The variant p. C31712R is the so far most common HMERF mutation [44]. The variant p. I35947N in the last exon is a well-known causative variant causing a dominant or a recessive disease as discussed below.
Functional data
The lack of scalable functional assays for TTN variants hampered a functional evaluation of the identified variants.
We considered TITINdb, a recently developed web application that maps titin variants to domain structures and computationally predicts their impact on the protein using both structure- and sequence- based methods, an excellent starting point to correctly localize the mutated amino acids [41]. For example, TITINdb clearly shows that the majority of the 59 re-occurring missense variants are located in fibronectin type III domains and it suggests a possible impact on the titin-obscurin and titin-obscurin-like interaction affinity for the re-occurring variant p.I35986T (mCSM score – 0.537 kcal/mol and – 0.777 kcal/mol, respectively).
Allelic and segregation data
Our preliminary results from segregation studies demonstrate that the interpretation of segregation data can be sometimes misleading as underlined in three examples below (Fig. 1): Dominant versus recessive effect: missense variants in exon 364 cause a dominant tibial muscular dystrophy (TMD) but, in combination with a second variant (in trans), they may cause several recessive phenotypes such as LGMD2J or a distal late-onset myopathy [2, 13]. In the family we describe in Fig. 1A, the proband carries the previously reported missense variant p.I35947N in exon 364 and a TTNtv. Although we have not confirmed the phase of these two variants because of the lack of paternal DNA, the disease is most probably recessive and due to the presence of bi-allelic titin variants. The sole identification of a variant in exon 364, although previously described, still requires a proper evaluation of its clinical effect through a segregation analysis and a careful clinical assessment (Fig. 1A). Pseudo-dominance: a specific variant (c.107647delT; p.(S35883Qfs*10) in exon363) co-segregated with adult-onset distal myopathyin an apparent dominant pattern in a French family (Fig. 1B) [45]. A critical re-evaluation of the clinical phenotype and a HTS-based re-analysis allowed the identification of two additional truncating variants in the affected mother and daughter [8], most probably suggesting a recessive mechanism. Reduced penetrance/variable expressivity: specific variants in exon 344 cause HMERF, an increasingly recognized phenotype [5, 42]. Although several cases with a dominant inheritance have been found, interestingly 2/10 of these causative variants were also present in asymptomatic carriers (Fig. 1C). Further studies are currently on going to clarify the possible effect of additional factors or secondary genetic hits to modify the penetrance and/or the expressivity of the primary mutation [5].
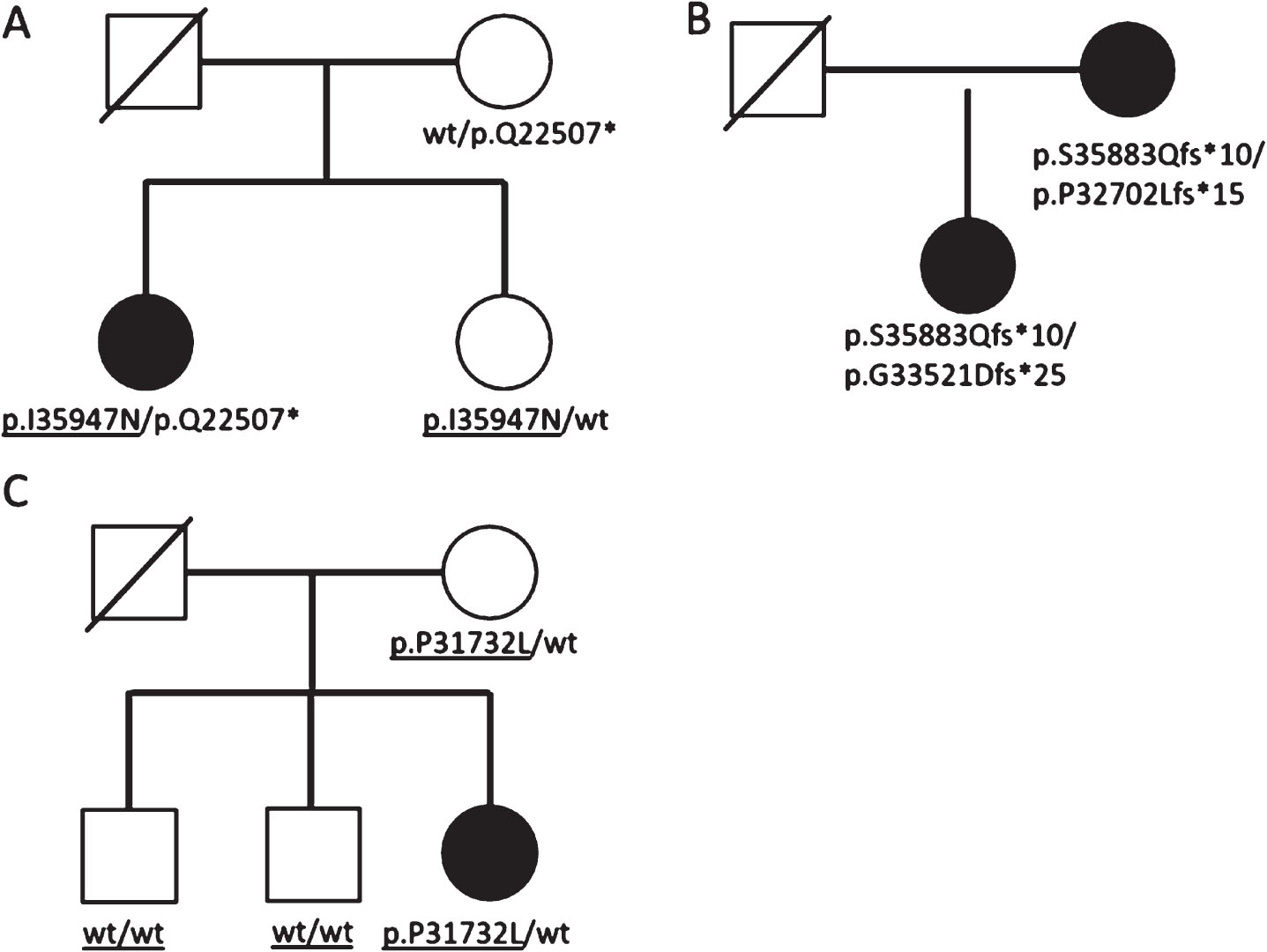
Segregation data. Segregation data in families with causative TTN variants can be sometimes misleading. A) Missense variants in exon 364 can cause either a dominant or a recessive phenotype. In the reported family, the phenotype in the proband is caused by the presence of the missense change and of a truncating variant (most probably in compound heterozygosity although the absence of sequencing data from father hampers the phasing of the variants). The sole, heterozygous missense variant may either be causative of a late-onset mild disease or may not be clinically relevant at all. B) The pedigree suggests the presence of a dominant disease due to a maternally inherited causative variant. On the contrary, three different TTNtv segregate in the family. The disease is recessive and, most probably, both the patients have bi-allelic TTNtv (the two TTNtv in the daughter have not been phased, however, clinical findings suggest that the variants are on two different alleles). C) The p.P31732L variant in exon 344, identified in the proband with an HMERF-compatible phenotype, is inherited by her asymptomatic mother, suggesting a possible reduced penetrance and/or the possible effect of secondary hits.
Probably disease-unrelated variants from TRIO- and DUO-based sequencing
We re-analyzed data from TRIO- and DUO-based tests, focusing on families with TTNtv, to identify probably benign variants.
We identified 14 variants (13 missense variants and one in-frame deletion) in trans with TTNtv in asymptomatic adult carriers (e.g. parents without a skeletal muscle disease) (Table 3). TTNtv have so far repeatedly been confirmed to cause recessive skeletal muscle phenotypes with or without cardiac involvement [2, 13]. Thereby, the 14 variants identified are most probably disease-unrelated.
Possible harmless variants
*Variants identified in trans with TTNtv in asymptomatic adults using TRIO and DUO-based sequencing tests.
DISCUSSION
The introduction of HTS technologies has revolutionized the approach to complex and genetically heterogeneous disorders, and made sequencing of large genes possible in patients [22, 46]. Exome and genome sequencing will probably soon be adopted as a first-tier test for all genetically heterogeneous conditions [22]. Genetic data may subsequently be available to clinicians during the first evaluation of a patient [22, 47].
Such extensive screenings will result in the identification of thousands of rare variants, which are difficult to interpret with current knowledge [12]. Simply because of its sheer size, variants in the TTN gene are detected frequently in any sequencing approach that includes this gene, whatever the clinical hypothesis might be.
TTN variants have been so far described in many different skeletal muscle and heart conditions [2, 48]. For a proper interpretation of TTN molecular findings additional investigations such as segregation studies, protein and cDNA assays are often mandatory.
The interpretation of truncating variants (TTNtv) in skeletal muscle disorders seems to be relatively straightforward. Their effects have so far always proved recessive, and bi-allelic TTNtv in a myopathy patient are most probably the cause of the observed phenotype [2, 13].
The interpretation of missense variants, which represent the vast majority of variants identified, is more challenging. Causative missense changes identified in myopathy patients seem to be responsible for both dominant and recessive skeletal muscle phenotypes.
Previously unreported TTN missense variants are to be considered variants of uncertain significance for which a pathogenic or predisposing role and a potential modifier effect cannot be ruled out nor confirmed.
With these considerations, how should we interpret missense TTN variants? Although the ACMG/AMP guidelines suggest evaluation of specific criteria [12], TTN and titinopathies are particularly challenging. Current predictive bioinformatic tools can be unreliable in predicting the effect of most missense TTN variants (Supplementary Table 1), and they can be very misleading in the clinical context. Moreover, although disease databases are a precious source of information for the assessment of the variants pathogenicity, currently a limited number of missense variants are listed (Supplementary Table 2). Finally, functional validations of missense variants in TTN are particularly challenging: the whole cDNA cannot be readily cloned and, so far, for in vitro testing, titin has been dissected into more manageable protein fragments [49]. These fragments can be expressed and purified to test a variant’s effect on protein–protein interactions or to verify its effect on the structure [49], but how relevant the results are to the effects of the variant in situ and in vivo is uncertain. Exonic variants causing missense changes can still affect the splicing. DNA-tests evidencing variants of uncertain significance should, if at all possible, be complemented by second-tier tests (e.g. RNA sequencing and protein analysis) to chase the presence of possible elusive molecular defects [50, 51]. Similarly, computational tools able to detect copy number variants and structural rearrangements should always complement, in a diagnostic setting, the traditional algorithms aiming at the detection of SNV or small indels [52]. Large TTN deletions have been identified in patients with recessive titinopathies [53].
We propose to refine the previous ACMG/AMP guidelines for a proper evaluation of TTN findings in the context of dominant and recessive skeletal muscle disorders (Table 4).
Modified ACMG/AMP pathogenic and benign criteria
HMERF = Hereditary myopathy with early respiratory failure, TMD = Tibial muscular dystrophy, SF = single family, MF = multiple families (>1)
Hallmarks of titin related skeletal muscle disorders
*Meta-only or metatranscript-only exons are those included in the metatranscript but not present within the six TTN isoforms described (N2A, N2B, N2BA, novex-1, novex-2, novex-3).
Quantitative adjustments
Four rules (PM2, PP1, BA1 and BS1) require disease-/gene-specific quantitative adjustments.
As already aforementioned, we consider a MAF<2.40E10-7 for variants causing a dominant skeletal myopathy and a MAF<9.80E10-5 for variants causing a recessive titinopathy a moderate indication of pathogenicity (PM2).
A MAF twenty times higher than the previously indicated thresholds (i.e. MAF > 4.80E10-6 for a dominant titinopathy or MAF > 1.96E10-3 for a recessive titinopathy) is an indication of non-pathogenicity (BS1). A MAF > 9.80E10-3 or > 2.40E10-5 for a recessive or dominant titinopathy respectively (i.e. one hundred times higher than the aforementioned thresholds) is a stand-alone proof of non-pathogenicity (BA1).
We are aware that setting MAF cutoffs for dominant and recessive titinopathies is a difficult task considering our current knowledge of these diseases. However, for a proper analysis, we do need to set plausible cutoffs based on a reasonable estimation of prevalence, penetrance, allelic and genetic heterogeneity, although we are aware that these thresholds may well be re-evaluated as our knowledge of these disorders, and their prevalence, evolves.
A large segregation analysis is important when assessing the pathogenicity of a previously unreported variant [54, 55]. However, the ACMG/AMP classification framework lacks a quantitative guideline for co-segregation criteria (PP1) [12]. Jarvik and Browning propose quantitative criteria for a strong, moderate and supporting co-segregation evidence [54]. The cutoffs they suggest are applicable also to titin variants and titinopathies [54].
Strength modification
In line with similar studies [27], we assign strong weight for pathogenicity/non pathogenicity only to functional evidence provided by a mammalian variant-specific knock-in model (PS3 and BS3). In vitro functional studies, performed using small fragments of the protein, can only support a pathogenic or benign effect (moderate evidence).
Similarly, the evaluation of splice-altering variants through RNA analysis or minigene splicing assays is crucial. However, if splicing variants cause an in-frame loss and result in a 'near full length protein' (e.g. due to the skipping of a symmetric exon), complementary protein and functional studies should be performed to confirm/exclude the pathogenicity of the observed variants.
The ACMG/AMP guidelines also suggest that the presence of variants identified in trans with a true pathogenic variant (for example a TTNtv) in recessive disorders is a moderate evidence of pathogenicity (PM3) [12]. However, the identification of a rare variant in compound heterozygosity with a TTNtv is a very common finding and the criteria can only be considered a supporting element.
Not applicable rules
We identified three rules not applicable to titin-related skeletal muscle disorders (PP2, BP1 and BP2). In particular, PP2 and BP1 are deemed not applicable because of the current partial knowledge of TTN disease-causing mechanisms.
Interestingly, ACMG/AMP guidelines consider the presence of variants in trans with a dominant variant or in cis with a pathogenic variant as a factor supporting non-pathogenicity (BP2) [12]. As clearly proved by either the HMERF semi-dominant variants (dominant variants with low penetrance) [5] or the aforementioned exon 364 changes [9], the presence of multiple causative variants in cis or in trans with dominant causative TTN variations cannot be excluded from having a disease causing role and a modifier effect on the phenotype.
Rules requiring clarification
Six rules (PM1, PP3, PP4, BS2, BP4, BP5), although still applicable, require a further clarification.
The location of a variant within a hotspot or a critical domain is a moderate evidence of pathogenicity (PM1). This applies to missense variants in exon 344, associated with HMERF [5, 42], and missense variants in exon 364, associated with TMD and/or recessive titinopathies with onset after infancy (proximal or distal myopathy) [2, 9]. It is also important to note that the location of TTNtv is crucial for a correct evaluation of a possible cardiac involvement (see reference [56] for an in-depth discussion of the role of titin variant in cardiomyopathy).
Moreover, we suggest that also data from in silico structural studies (e.g. data from TITINdb [41]) can be considering supporting evidence of pathogenicity or non-pathogenicity along with prediction from bioinformatic programs (PP3 and BP4).
The rule PP4 mentions the phenotype as possible supporting evidence for variant claims. However, as clarified in the guidelines, phenotypic elements provide supporting evidence only in presence of a well-defined disease distinguishable from other clinical entities. Although an exhaustive definition of titin-related clinical symptoms and signs is beyond the scope of this study, a combination of clinical, histopathological and imaging features can address the diagnosis. (Table 5) [4, 13]. A larger consensus on the diagnostic and prognostic value of TTN-related features is however advisable and will improve a standardized interpretation of TTN findings.
The rule BP5 suggests that the identification of a gene variant in patients with a different confirmed molecular diagnosis (i.e. with other mutations explaining the observed disease) can be considered a supporting evidence to classify that specific variant as benign. As also suggested in the guidelines, BP5 application is more straightforward with monoallelic variants and possible dominant diseases. Considering our current partial knowledge of TTN-related diseases, we suggest this rule to be only applied to patients with a clinical condition not involving cardiac and skeletal muscles. Moreover, the age of patient at the genetic examination must be carefully evaluated. For example, the identification of a missense variant in exon 364 in an infant with epilepsy does not suggest that the variant is benign (heterozygous missense changes in exon 364 may result in very late adult onset titinopathies). Vice versa, the identification of a heterozygous mutation in exon 344 or 364 in an elderly patient with, for example, familial adenomatous polyposis without any respiratory and muscle symptoms/signs suggests that the variant does not cause a dominant skeletal muscle disorder.
Finally, for recessive titinopathies, variants identified in a healthy adult have an evidence of a benign impact if identified in homozygosity or in trans with a variant causing a premature stop codon (BS2).
CONCLUSIONS
We suggest specific adjustments to the ACMG/AMP variant classification guidelines useful for a proper interpretation of TTN variants in the context of skeletal muscle disorders. We expect that the rules we suggest here will be further improved along with a better understanding of TTN-related skeletal muscle disorder.
At the same time, our work provides a proof of principle of the benefits of a possible collaborative effort among all the different specialists working on titin aiming to improve the interpretation of TTN variants using a large cohort of patients. The integration of deep phenotypes on a per-patient level, and computerized data sharing mechanisms are essential [57] and strongly advocated by the rare disease community [58, 59]. Increased sharing of genetic and clinical data would greatly improve our current understanding of TTN-related diseases, enabling a refined burden analysis of titin variants.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
Footnotes
ACKNOWLEDGMENTS
The authors would like to thank all the patients and family members for their cooperation; all the clinicians for collecting patient data; Merja Soininen, Helena Luque at Folkhälsan Research Center and Gaia Esposito at TIGEM for technical help; Tiina Suominen and Antti Väisänen at Neuromuscular Research Center (Tampere) for acquisition of samples.
This study was supported by Association Francaise contre les Myopathies (M.S.), Orion foundation (M.S.), Magnus Ehrnrooth Foundation (M.S.), Päivikki ja Sakari Sohlbergin Säätiö (M.S.), Jane and Aatos Erkko Foundation (P.H.), Medicinska Understödsföreningen Liv och Hälsa rf (P.H.), Folkhälsan Research Foundation (B.U.), Erkko Foundation (B.U.), Juselius Foundation (B.U.), Finnish Academy (B.U.), Telethon Italy (V.N.) and Telethon-UILDM (Unione Italiana Lotta alla Distrofia Muscolare) (V.N.). The MYO-SEQ project was supported by Sanofi Genzyme, Ultragenyx, LGMD2I Research Fund, Samantha J Brazzo Foundation, LGMD2D Foundation, Kurt+Peter Foundation, Muscular Dystrophy UK and Coalition to Cure Calpain 3. The HTS work in inherited myopathies in Pisa Lab is supported by Regione Toscana FAS SALUTE 2014 (CUP 4042.16092014.066000060 to FMS). H.L. and R.T. are supported by the European Union Seventh Framework Programme (FP7/2007-2013) under grant agreement Nos. 305444 (RD-Connect). AM is supported by Intramural Research Program at National Institute of Neurological Disorders and Stroke.